{ version = 15.38; (* of lister.p 2025 Aug 21}
(* begin module describe.lister *)
(*
name
lister: list the sequences in a book with translation and sequence walkers
synopsis
lister(book: in, listerp: in, features: in, colors: in, marks: in,
list: out, map: out, output: out)
files
book: Any book generated by the Delila system.
listerp: Parameters to control the listing. If listerp is empty, default
values are used. Otherwise, the file must contain the following
parameters, one per line:
1. The version number of the program. This allows the user to be
warned if an old parameter file is used.
2. basesperline: the number of bases per line in the listing. Note that
besides margin characters, there will be one blank with each base.
This must be a multiple of 3 whenever one is printing amino acids.
3. aastate: the mode for listing amino acids:
0 = none
1 = predict peptides starting at aug, gug or uug.
show nonsense codons. uug is new as of 2007 Mar 16.
2 = translate all frames
In the special case that predicted reading frames starting at
identified ribosome binding sites are emphasized, the
predicted peptides outside the emphasized frame are
suppressed. To get these anyway, use the negative value of
this parameter. That is, use aastate = -1 or -2. (The
variable is called giveframes.)
4. frameallowed: an integer in the range 0 to 7. The binary
representation of this number determines which amino acid frames are
allowed to be printed. The highest bit is the highest printed frame.
5. codelength: Amino acid code: one character
1 = 1 letter code
3 = 3 letter code
6. seqlines: an integer that controls the listing of the sequence.
0 = no sequence (but show amino acids and sequence numbering)
1 = show sequence
2 = show sequence and complement underneath
7. pageaction: one character
c = computer defined page character (often will be a control-L)
l = LaTeX document page notation
n = no page marks
8. pagelength (integer): the number of lines per page.
This will determine when lister starts a new page. When displayed
by ghostview, it has the nice property of showing only the
number of lines given, so the display can be 'shrinkwrapped' around
the interesting features. See the pageedges parameter.
9. "hopi": Header control: The first 4 characters of the line are "hopi"
(in that order). These define the parts of the list to show:
header: version number program name, etc.
organism/chromosome name
piece information
index
The characters are either lower case (do not display) or upper case
meaning to display. For example, hoPi would only give the piece
information. Special case: if the first letter is 'B' then only
the book date and title will appear in the header.
The next 4 characters on the same line, NKFC, control the
parts of the piece information to display:
N: number (the word 'piece' will proceed it)
K: key name (eg, GenBank accession)
F: full name (this can be controlled by the Delila name command)
C: coordinates of the piece
As with hopi, only upper case letters turn on the option.
10. "ndaf": data control: The first four characters of the line define
the lines of data to show. The possible characters are:
N show numbering of DNA sequence
D show DNA sequence
A show amino acid sequence
F show features
These may be presented in any order. Capital letters turn on the
feature, small letters turn it off. No other characters are
allowed.
11. "sof": feature position control: three characters on the line.
If the first character is an upper case 'S' (same line) then the
features that run from one line to the next are always the SAME
distance from the sequence. Reasoning: For each line of the
sequence the features are written in the order they are found in
the features file. When there is space available, features are put
closer to the sequence. If a feature continues on the next line,
it will often be a different distance from the sequence. One way
to avoid this would be to make a long line and then use the split.p
program. This parameter avoids that step. Otherwise, the features
are placed as close to the sequence as possible.
If the second character is an upper case 'O' then only ONE feature
is put on each line. Note that this alone does not guarantee that
features will remain on the same line.
WARNING: if a plot is very complicated, the ellipsis' on one line
will not be in the same order as the next line. In these cases it
is better to use 'S' or turn on 'O'.
If the third character is an 'F', then features are placed in the
order ("FORWARD") given in the features file. If this character is
an 'R' features are placed in the REVERSE order. (This is
implemented simply by reversing the list.) To see the effect of
this, consider a series of features that saturate the sequence, one
every base. For simplicity, set the ONE feature per line mode on
(see above). In the Forward mode, the first feature would appear
on the "upper left" and successive ones would appear towards the
"lower right" as in the backslash "\". In the Reverse mode, the
first features would appear on the "upper right" and proceed to the
"lower left" as in the slash "/".
12. pagetrigger: Pages are normally started when the list goes beyond
the pagelength. If the first character on this line is 'p' then a
new page will start every time a new piece of DNA is encountered in
the book (actually it will when the organism/chromosome information is
to be put out). If the first character is 'o' or 'c' the trigger will
be at the organism or chromosome changes, respectively. Other
characters leave triggering at the end of the normal page.
The action take at the end of the page is determined by pageaction.
If the pagetrigger is 'l', then the next lines list the numbers of
pieces in the book that should trigger paging. This allows
arbitrary numbers of pieces per page, under program control.
The list is started with a delimiter number. This number must
be also at the end of the list. For example if there are no
pieces numbered 0, one could make the list with "0 3 5 7 0",
which would put pieces 1 and 2 on the first page, 3 and 4 on
the second page, etc.
Doubling: if the pagetrigger is 'd', paging occurs on odd
piece numbers. This is the same as l but without the list.
The equivalent l list would be like: "0 3 5 7 0".
Capital letters for any of pocld will prevent page numbers but
otherwise will page as before.
To prevent page triggering, use a space.
To make no page number on the first page use N.
13. map control: A series of values:
* mapcontrol: If the first character on the line is a 'C', then the
color map file will be written. If it is 'R' then the page will
be set up so that the upper left corner is moved to the lower left
corner and the image is rotated 90 degrees counter clockwise.
This has the effect of making the image in "landscape" mode.
* fontsize (integer): The character height in points (there
are 72 points/inch, 2.54 cm/inch). Typical values: 10 or 12.
14. image positioning controls
* deltaXcm: The amount to move the image in X (cm).
* deltaYcm: The amount to move the image in Y (cm).
* scaleimage: the scaling factor.
The image will be shifted on the printed page. X is positive to the
right and Y is positive up the page. Generally one would use
positive values for X and negative values of Y since the image
should otherwise fit snugly in the upper left corner of the page.
When the "shrinkwrap" mode is on (see parameter pageedges below) the
position will have no effect on viewing by GhostView but the image
will still move around on the printed page. This is quite
convenient because it means that one can position an image on the
printed page as one would like independently of the display on the
computer screen.
The scaling is performed after movement from the upper left hand
corner of the image as one would read it. If the image has been put
in "landscape" mode the delta-shifts are given in the new coordinate
system. This allows one to switch between "landscape" and regular
"portrait" mode without changing the parameters, and it allows one
to think in terms of a normally held page.
15. lowerbound: The lower bound of the walker display box, in bits. It
must be negative. See figurebottom.
If a base goes below lowerbound, Lister clips the letter to
lowerbound and puts a purple box.
If a base has never been seen before at a position in a binding
site, then Lister shows a black box.
16. figurebottom: How far down the bottom of the walker display is, in
bits. It must be negative. See lowerbound.
Lowerbound is for displaying the box at the zero coordinate of the
walker, while figurebottom controls the number of lines the bottom of
the walker will take up (1 line = 1 bit). By separating these two
functions the user has more control over the display.
17. shiftsequence (integer): The sequence will be shifted to the right
by this many bases. It must not exceed basesperline. Shifting left
is done with Delila instructions by removing sequence. This allows
one to make figures with the sequences lined up as one would like.
18. pageedges pageboundaries extraright (character, character,
integer): The size of the map page shown in GhostView is
determined by these parameters.
pageedges (character):
If the first character is 'd' then default values are used that
show the region that will be printed on a standard printer. See
technical notes for further details.
If the first character is 's' then the program will "shrink wrap"
the page to fit the parameters basesperline and pagelength.
pageboundaries (character): If the second character is 'b' then the
program will show the default page boundary as a red line running
around the page. If any graphics touches this red line, it will
not appear on the printer.
extraright (integer): Since header lines and features can extend to
the right, the integer defines the number of extra characters to
allow for on the right side of the page.
19. edgecontrol edgeleft, edgeright, edgelow, edgehigh:
edgecontrol is a single character that controls how the bounding
box of the figure is handled. If it is 'n' then the bounding box
will be the page parameters defined in constants inside the program
(llx, lly, urx, ury AND changes as set by the previous parameter
line). If the parameter is 'p', there are four real numbers that
define the edges around the clist in cm. To allow a map to be
imbedded into another figure, its size must be defined in
PostScript (with %%BoundingBox). By setting these four numbers,
the edges are defined. Negative values are allowed, so one may
move the edges as desired.
These parameters are the same form as those in the alist program.
The definitions are IN ADDITION to the parameters on the previous
line. Thus pageedges d and s on the previous line still give the
default page and the shrink wrap. The extraright in the previous
parameter line is in CHARACTERS so that one may control the right
edge precisely. By contrast, the edgeright of this parameter line
(and all others) is in cm.
(New as of 2001 Aug 29)
20. KeepDuplicates (character): if the first character on the line is a
'k', duplicate features will be kept. If not, duplicates will be
eliminated. One use of duplicate features is in a "supporting"
role: by putting a feature that prints as a blank space in the
location of a visible feature, that visible feature will be forced
further down the page. This gives one more control over the
placement of features, but if one is using a lot of supporting blank
features, then they must not be eliminated by the program as
duplicates.
Duplication is defined as having the same id, position and
orientation.
21. Sideways (character): if the first character on the line is an 's',
asymmetric walkers will be shown with sideways characters. The
direction of the walker is the direction that one would read the
letters "down the page".
22. MinimumLines (integer): This number defines the minimum number of
lines between sequences. It is intended to be used in cases where
one wants to compare a page of sequence to another page that has a
slightly different sequence. Features will appear or disappear and
so normally the number of lines used will vary. This is not good if
one wants to make a movie or compare two sequences. By setting
minlines high enough, one can ensure that the locations of the lines
will not vary.
23. artmode (character): if the first character on the line is
an 'a', then all black ASCII characters on features are
removed. This mode has become possible because features can
now be displayed with sequence walkers and colored boxes for
ASCII features. If the character is 'n' then names will be
given and objects that have no color will be given. If the
character is 'b' the art mode is activated and the background
becomes black. One pixel is provided around the standard
boarder (llx, lly, urx, ury). If the character is 'B' the
art mode is activated and the user can use the edge control
to change where the black is.
24. outline (character), outlinewidth (real) outlinegray (real)
If the first character is 'o' then the letters of each
sequence walker will have an outline around them.
The second parameter determines the stroke width. 0.0 will
give 1 pixel wide (as defined in PostScript). 1.0 may be too
wide, but real values in between, such as 0.3, seem ok. The
letter is clipped, so only half of the stroke width is
actually drawn. If one uses a large value, then the entire
letter becomes filled in, providing a way to make walkers
that are entirely shades of gray.
The third parameter is the gray scale value to use for the
outline. 0 is black and 1 is white. (There is no option for
color.)
The reason for allowing this option is that when petals are
used, a very strong site can have a petal rectangle that is
the same color as a sequence walker letter. For example, the
default petal color is red and the default color for the base
t is also red. When the petal is under a strong binding
site, all of the t's are invisible against the petal
background. By making an outline, the letters can be
distinguished from the background, but at the expense of them
becomming thinner. Beware that with some printers it may not
be possible to see outlines that are too thin.
(New as of 2005 Nov 9.)
25. horizontal (character) symbol (character) width (real) height (real)
If the first character is h, then provide a colored bar
for the Gap connection lines.
The second character, the symbol for the horzontal line is
ignored. It is there only for consistency with the vertical
control. The feature symbol is controlled in the feature itself
and this does not override.
The width and height are scale factors from a standard
colored bar. Leave them as 1.0 and 1.0 to get the standard.
Example:
h+ 1.0 1.0 horizontal: h(orizontal bar) - width height
26. vertical (character) symbol (character) width (real) height (real)
If the first character is v, then provide vertical colored
bars in multipart walkers.
The second character is the symbol for the vertical line. If
you set it to space (' ') then there won't be one.
The width and height are scale factors from a standard
colored bar. Leave them as 1.0 and 1.0 to get the standard.
Example:
v| 1.0 1.0 vertical: v(ertical bar) | width height
features: A list of locations of binding sites and other features. Lines
that begin with "*" are comments. Completely blank lines are
skipped. Otherwise each line defines the location or properties
of a site.
* comment
d definition of a feature
r ribl matrix
b boundary for site
c color rectangle
@ location of a feature
g beginning of a multi-part group of features
For example, to generate
*260 * *270 * *280 * *290
5' t t a t c c a c a g t a g a t c g c a c g a t c t g t a t a c 3'
[------*------0-----^---> Site Positive 270
<---v-----0------*------] Site Negative 270
[------*------0-----^---> Site Positive 280
one uses the four lines:
define "Site" "-" "<v0*]" "[*0^>" -7 -3.5 0 +3.0 +5
@ K01789 270 +1 "Site" "Positive 270"
@ K01789 270 -1 "Site" "Negative 270"
@ K01789 280 +1 "Site" "Positive 280"
The first line defines a site called "Site".
"-": use dashes to pad out the feature background.
"<v0*]": the characters to use in the negative orientation
"[*0^>": the characters to use in the positive orientation
-7 -3.5 0 +3.0 +5: the location of the characters relative to the zero
coordinate of the binding site.
The background string must contain only 1 or 2 characters. The
character mark strings may be any length (up to a limit defined in the
program), must be the same length, and that length must correspond to
the number of numbers that follow.
If the second character in the background string is "d", then the
background will be the DNA sequence and the spaces between the
sequence will be the first character. The complementary base is given
when the feature orientation is negative. Note if one puts marks on
the feature at the half base positions, they will not interfere with
the sequence.
If the second character in the background string is "w", then the walker
will be displayed. This consists of characters with varying heights that
represent the sequence conservation of the given base, measured in bits.
A "W" will show the bounding box on each walker character. The
complementary base is given when the feature orientation is negative.
If this character is l, it acts as "w" but shows lines at integer
values of the bits.
If this character is L, it acts as "W" but shows lines at integer
values of the bits.
If this character is i, it acts as "w" but shows lines at integer
values of the bits only on the walker.
If this character is I, it acts as "W" but shows lines at integer
values of the bits.
The second line in the example above (beginning with "@") defines the
location of a binding site on sequence K01789. The zero of the site
is placed at coordinate 270. The feature is to be oriented in the
positive direction relative to the given sequence. The name of the
site is "Site". Additional information to be listed about the site is
"Positive 270".
The site will be marked every time it is found, even there are
multiple copies of a piece by the same name in the book. Sites which
extend outside the line are listed with the ellipsis "..." to indicate
continuation.
Tabs inside quoted strings are converted to blanks when they are
written out. This allows the tab to be a special "solid" kind of
blank, but the string written won't vary with position on the line
depending on the tabstops. The program will also accept blanks at the
ends of lines. These are converted to tabs internally. These
features allow one to extend the length of a feature to the right so
that it will exclude other features from that area.
When features are generated automatically by the scan program, the Ribl
matrix is stored in the features file. This begins with the letter 'r'
followed by a quoted string (eg "Fis") that relates the matrix to the
definition. This is followed by the standard ribl file format defined in
the ri program. These data allow the walker to be created, but since it
is done automatically the user usually need not create this by hand.
If the feature has an Ribl matrix definition (because, for example it was
created by scan as described above), then the three values for Ri, Z and
probability follow the "other" string. These are usually generated
automatically by the scan program and not by the user.
As in the walker program, the walker has a light green or pink
bar at zero. The color of the bar is determined for each feature
from the evaluation values mentioned above and from new boundaries
defined by the user:
* Ribound: lowest Ri for producing a green bar
* Zbound: lowest absolute Z score for producing a green bar
* Pbound: lowest probability for producing a green bar
These cannot be defined in the scan program since one would like to scan
with lenient bounds and then make a more stringent bound at the lister
level to mark those which passed the second stage. Thus strong sites are
shown in green and weaker ones in pink. This requires a definition of
the boundaries. The first character of the line must be 'b'. The rest
of the word is ignored. This is followed by a quoted string that relates
the boundaries to the definition. The three bounds are then given. An
example for the Fis features is:
boundaries "Fis" 10.5 2.0 0.20
where Ribound = 10.5, Zbound = 2.00 and Pbound = 0.2. This means that
only sites with Ri> Ribound, Z <= Zbound and probability >= p will get a
green bar. (The order of these paramters is the order they
are computed in. Ri determines Z, and this determines probability.)
NO LONGER TRUE:
Any characters on the line after those expected (the list of locations
for definitions and the second quoted string for the features and the
Ri, Z and probability) are
ignored and may be used as comments.
COLOR OF CHARACTER FEATURES
The color of text character features can be changed by adding
a set of 5 parameters at the end of the '@' line.
Tparam: char; a parameter that defines the kind of parameter
that the remainder parameters are.
Aparam: real; a parameter
Bparam: real; a parameter
Cparam: real; a parameter
Dparam: real; a parameter
Tparam values:
h = hsb feature colors: hue, saturation, brightness
r = rgb feature colors: red, green, blue
b = bit range for the feature
' ' = defaults to sequence walker
Summary:
Tparam Aparam Bparam Cparam Dparam For
' ' Ri Z P (none) walker
'h' hue saturation brightness (none) ascii lines
'r' red green blue (none) ascii lines
'H' hue saturation brightness thickness color bar
'R' red green blue thickness color bar
In this scheme one can control the color of the bar as an
ascii string OR one can control the colored rectangle behind
the bar but NOT BOTH.
Many feature files may be concatenated together to create complex
features. This allows the user to add their own features and to set
the boundaries as defined above.
If a program that generates features searches a book with several
identical pieces, then multiple copies of a feature can be found.
These duplicates are removed by the program. (To allow the duplicates
to be kept, the parameter KeepDuplicates can be set to true.)
Note that the program cannot identify duplicates if they are on
the opposite strand!
New as of 1999 March 11:
Makelogo has the ability to put cosine waves on the sequence logo.
This is now possible for lister walkers. If the ri program is run with
a non-empty wave file, then the ribl will contain a wave definition.
This is passed into the scanfeatures file and and used to mark the
walker. Thus if one defines one (or more!) waves on the sequence logo,
these will automatically appear on the corresponding walkers!
New as of 1999 April 1:
One can have features with italic characters in them. For example to
make the word 'Italy' be in italics, use the string:
\)\(Italy\)IT\(
This can be done both inside the Delila instruction name string and
inside the names of features. To use it in the search strings, one
must replace all spaces by tabs, but otherwise the syntax is the same.
TECHNICAL NOTE (how it works): The first right parenthesis closes any
previous postscript string. The word to be italicized is in
parenthesis. Then the IT function is called to write it out. The
final left parenthesis starts any remaining postscript string.
WARNING #1: Other features on the rest of the line may be displaced
slightly.
WARNING #2: You must use tabs instead of spaces if you generate your
features using the search program. You can use spaces in delila name
instructions.
New as of 1999 April 7:
One can generate any of the standard PostScript symbols (PostScript
Language Reference Manual, second ed, 1990, ISBN 0-201-18127-4, section
E.11, Symbol Character Set, p. 604). For example, this will produce a
beta symbol: "\)\(\\142\)SY\(". Note that two backslashes are needed
in front of the 142 to ensure that a backslash appears in the
PostScript.
New as of 2003 Aug 18:
If Delila halts, then the scan program halts and an 'h' appears
in the feature file. This is now a valid initial character
for features, and it triggers a warning.
COLOR RECTANGLES BEHIND WALKERS: petals *********************
New as of 2004 July 13
Color rectangles behind walkers - called 'petals' can be
defined. This allows one to see each kind of walker with its
own color. The form of the line is:
petal "ribl_name" el c eh es eb fh fs fb th
ribl-name = the name of the kind of walker to use
el = edgelinewidth: edge linewidth (integer)
c = color kind: how color is defined: r for RGB, h for HSB
eh = edgeh: edge hue OR red
es = edges: edge saturation OR green
eb = edgeb: edge brightness OR blue
fh = fillh: fill hue OR red
fs = fills: fill saturation OR green
fb = fillb: fill brightness OR blue
th = thickness of the petal
The user defines:
- the name of the Ribl weight matrix (inside quotes);
- the edge thickness (el); If el is 1 or larger, then el is the
thickness in PostScript points (1/72 inch). If el is 0 then the
thickness is 1 pixlel on the display. If el is negative then no
edge is given.
- whether to use
RGB (red, green, blue) or HSB (hue saturation brightness)
for the color definitions (values range between 0 and 1);
- the color of the edge of the rectangle;
- the color of the interior of the rectangle;
The thickness is a number between 0 and 1.
When it is 1.0, the height of each colored rectangle
is the full height of a character in the font.
As this number decreases, the rectangle shrinks in the
vertical direction to the center of the character.
These parameters apply only to the walker defined.
The ribl must be defined before the petal definition.
Color Intensity Control:
The color values can be controlled automatically. The Ri value
of the site is known and the consensus is known. Dividing Ri by
the consensus gives a range from 0 to 1. If the Ri is negative,
0 is used. Color values (any of hue, saturation, brightness,
red, green and blue) are normally in the range 0 to 1, so to
trigger this mechanism for any color value, use a value less
than zero. The color value will then vary with the information
content of the site.
When the color value is negative, use a value of -1 to
get the range 0 to Ri/consensus.
In general, when the color value is negative, use other values
to scale up the color range. That is, if the value is scale < 0,
then the color range will be 0 to Ri*abs(scale)/consensus. Any
values that exceed 1 are set to 1. This allows one to enhance
the sensitivity.
The most simple way to vary the color is to use the hsb model
and to have the hue determined by the binding site model, the
brightness fully up and the saturation determined by the
information content of the site. Thus a red site with zero
information content will be white, one with about average
information content will be pink and the strongest site (the
consensus) will be full red. The mkpetals script will
automatically set this for you if you provide it with a features
file that contains ribl files (the usual output of scan or
multiscan). In PostScript, the first color is always red so the
output for the first sequence walker in the features file are
called "rose petals".
*************************************************************
colors: colors defining the bases, see makelogo for definition. Each line
either begins with a '*' (comment) or a character. The character is then
followed by three real numbers (between 0 and 1) that define the amounts
of red, green and blue. There is also a conflict between the amino acid
letters and the DNA sequence bases, but is be resolved by making the
bases only in lower letters case and the amino acids only in upper case
letters in the colors definition file.
marks: Define the location of marks on the map. See makelogo.p for
details of this method. Note that the horizontal positions are given in
coordinates of the piece (rather than coordinates relative to the zero
base of the logo) and that vertical positions are still given in bits,
with one bit per vertical character size. The marks are relative to the
first line of the listing, so mark positions should usually have negative
y (bit) locations. These marks are global and appear under all other ink
put on the page. They should not be confused with the character marks of
individual features. Marks are drawn before the other features in the
listing and so appear underneath them. Marks are drawn from the left to
the right and the first coordinate of a mark defines its order of
drawing. The order of the marks must be the order they are to be drawn
in. As described in makelogo.p, the user can define their own marks.
SPECIAL COMMAND: p
There are many times when one would like to skip ahead to the next
piece in the book for the next mark. The 'p' command in the marks file
(ie, just p at start of line with any characters following) means that
marks should not be gotten until the start of the next piece. By
convention, one does not need to step to the first piece, and one
should always use the p command to have lister move to the next piece
when one is done with a given piece. These rules prevent insanity.
SPECIAL TRICKS:
One can redefine Postscript functions in the marks file if one knows
how lister works. In particular, if one needs to make a black and
white figure, then the black down box will be the same shade as the
base and the base won't be visible. To allow the normally purple and
black down box colors to be changed, both are defined as 'downblack'
and 'downpurple'. These can be redefined. For example: "/downblack
{0.9 setgray} def" redefines downblack set the shade to gray so that
the base is not wiped out.
writeln(output);
SPECIAL COMMAND: o, which controls the marksorder parameter. The 'o'
command controls when the marks are written. For example, it allows
one to overwrite the DNA sequence with marks. The "o" command must be
followed by a character, bNADF. The default is that marks are written
BEFORE (b) everything else, so they are under all other graphics. The
other values set the time that marks are written: N (after the
numbers), D (after DNA), A (after amino acids) or F (after features).
Note that the coordinate system changes after each of these: it is
displaced downwards.
list: A carefully numbered listing of the sequences in book, with amino
acid translations and features marked and with an index to the pieces
at the end. The file is pure ASCII.
map: like list, but the sequences are in color, according to the colors
given in the colors file. The file is in PostScript graphics. If the
definition of a site is marked as a walker and if there is a ribl
information, then the walker will be displayed.
output: messages to the user
description
Lister is a general purpose program for the listing of nucleic-acid
sequences. Bases are separated by one space. This gives enough room for
many features to be shown.
The listing can have numbering in which every fifth base is carefully marked
with an asterisk directly above it. Every tenth base is numbered with the
number defined by the coordinate system.
The listing can include translation to amino acids. The amino acid is set
directly below the codon. Dashes mark the frame.
The listing can include various features defined by a user or automatically
found by another program.
The listing can have various user defined marks placed on it. The locations
of these marks are given in convenient coordinates (bases and bits).
Walkers are shown on the color listing (map). The location to place the
walkers is determined by the scan program, and read into this program by
the features file. To allow concatenation of several scans together, the
output of the scan program is called scanfeatures. Walkers consist of a
set of letters. The zero base is shown by a rectangle. The upper line of
the rectangle is at 2 bits, the lower line is given by the figurebottom
parameter. One line of the display is equal to one bit. The zero height
is indicated by placement of the letters as described below. The letters
of the walker indicate several kinds of data. Their height is in bits.
If they point downward, then that represents a negative contribution of
that base to the individual information. The orientation of the letters
indicates the symmetry of the site. There are three possible symmetries:
asymmetric, even and odd. A site with odd symmetry has a base in the
center. A site with even symmetry has a phosphate in the center. The
symmetry is determined by the ri program, and passed to the scan program
in the ribl file. When a site is symmetric, the walker letters are
vertical. In this case if their contribution is negative, the letter will
be rotated 180 degrees so it points downward. If a site is asymmetric,
then the walker letters are rotated -90 or +90 degrees (when the Sideways
parameter is set to 's'). The direction of the site is the direction one
would read the letters if they were running down a page of text. If the
base contributes negatively to the Ri, it is below the zero line of the
walker.
examples
If listerp contains:
11.55 version of lister that this parameter file is designed for.
38 basesperline: number of bases per line in the listing
1 aastate: 0=no aa; 1=predict peptides; 2=translate all frames
7 frameallowed: binary; highest bit is highest frame on, etc.
1 codelength: 1 or 3 letters per amino acid
1 seqlines: 0=no sequence; 1=single strand; 2=double strand
c pageaction: c=computer; l=LaTeX; n=none
60 pagelength: page length
hoPiNKFC hopinkfc: Header, Organism/chromosome, Piece, Index; #, key, full, coords
NDAF ndaf: Numbering, DNA, Amino acids, Features
SoF sof: feature position, S=on SAME line, O=ONE per line, FORWARD/REVERSE.
p pocld: pagetrigger, piece, organism, chromosome; list, double
C 10 mapcontrol: C=do color map, R= rotate, char height (72 points/inch)
0.3 -0.3 0.5 amount to move image in x and y (cm) and scale factor
-3 lowerbound (bits) lower bound of walker box graphic
-3 figurebottom (bits) lower bound of walker in lines
0 shiftsequence (bases) amount to shift the sequence to the right
sb 20 pageedges: d(default), s(shrinkwrap); pageboundaries (b/n); extraright (integer)
n 0 0 0 0 edgecontrol (p=page), edgeleft, edgeright, edgelow, edgehigh in cm
not keep KeepDuplicates: k(keep), n(o): keep (or not) duplicate features
sideways Sideways: s(ideways), n(o): show walker features sideways
0 MinimumLines: minimum lines between sequences
- artmode: a(rtmode), n(ames), b(black), B(black, controlled), otherwise not.
o 0.3 0.0 outline outlinewidth outlinegray
this means:
11.55 this parameter file will not work with versions less than 11.55
38 the listing will be 38 bases wide
1 with predicted peptides for
7 the top frame.
1 The translated sequence is listed in single letter code.
2 Both DNA strands will be given.
c The computer's default will be used to page the output.
60 Each page will break at 60 lines.
h no header will be shown
o no organism/chromosome information will be shown
P the information about each piece will be shown
i no index will be printed at the end.
N show piece number
K show piece key name
F show piece full name
C show piece coordinate system
N show Numbering of DNA sequence
D show DNA sequence
a don't show Amino acid sequence
F show Features
S features are kept at the same level
o more than one feature is printed per line
F features are in the order given in the features file
p trigger a new page at every new piece.
C C = do map
10 character height 10 points
0.3 move image 0.3 cm x on the page (ie right)
-0.3 move image -0.3 cm y on the page (ie down)
0.5 make the image half size
-3 the walker boxes will go down to -3 bits (lines)
-3 the walker will take 3 lines (bits) below zero
0 The sequence will not be shifted.
s When viewed with ghostview, the image will just fit inside the viewer
b The edge of the printable page will be shown in red
20 the right edge of the image will be extended 20 cm
n edgecontrol -none (p=page will be controlled)
0 edgeleft: additional edging in cm
0 edgeright: additional edging in cm
0 edgelow: additional edging in cm
0 edgehigh: additional edging in cm
not don't keep duplicate features
sideways rotate the asymmetric walker letters
0 no minimum to lines between sequences
- artmode turned off, black ASCII characters will be shown
o outline: letters will have outlines
0.3 outlinewidth: half of the width in points (1/72 inches)
0.0 outlinegray: 0 is black, 1 is white for the outline
More examples for frame control (parameter 3):
7 (111 in binary) will translate all frames
4 (100 in binary) will translate only the first frame
3 (011 in binary) will translate the second and third frames
More feature examples
define "GATC" "-" "[0*]" "[*0]" -1.5 -0.5 0 1.5
@ K01789 281.5 +1 "GATC" ""
@ K01789 281.5 -1 "GATC" ""
define "GAnCT" "-" "[0*]" "[*0]" -2 -1 0 2
@ K01789 282.0 +1 "GAnCT" ""
@ K01789 282.0 -1 "GAnCT" ""
produce:
*260 * *270 * *280 * *290
5' t t a t c c a c a g t a g a t c g c a c g a t c t g t a t a c 3'
3' a a t a g g t g t c a t c t a g c g t g c t a g a c a t a t g 5'
[-*0--] GATC
[--0*-] GATC
[-*-0---] GAnCT
[---0-*-] GAnCT
Sometimes one would like to force a particular feature to be somewhere
else. One could do this by putting a visible feature there, but then one
would have characters one does not desire. A trick is to define a totally
blank feature:
* define an invisible feature to displace other features
define " " " " " " " " 0
@ J02459 27731 +1 " " " "
This feature will displace other features, but since it has only blanks, it
will not show up itself! By putting this feature before other features in
the feature list, it will get priority for positioning.
COLOR OF CHARACTER FEATURES
define "PEAK" "-" "<0]" "[0>" -1 0 1
@ NC_000913 152.0 +1 "PEAK" "" R 0 1 0 1
This makes a green colored '[0>' one character high.
documentation
@article{Schneider.walker,
author = "T. D. Schneider",
title = "Sequence Walkers:
a graphical method to display how binding proteins
interact with {DNA} or {RNA} sequences",
journal = "Nucleic Acids Res.",
volume = "25",
comment = "walker.tex, November 1, issue 21",
note = "\htmladdnormallink
{https://alum.mit.edu/www/toms/paper/walker/}
{https://alum.mit.edu/www/toms/paper/walker/},
erratum: NAR 26(4): 1135, 1998",
pages = "4408-4415",
year = "1997"}
@article{Shultzaberger.Schneider2001,
author = "R. K. Shultzaberger
and R. E. Bucheimer
and K. E. Rudd
and T. D. Schneider",
title = "{Anatomy of \emph{Escherichia coli}
Ribosome Binding Sites}",
journal = "J. Mol. Biol.",
volume = "313",
pages = "215-228",
comment = "Shultzaberger.Schneider.flexrbs",
note = "\htmladdnormallink
{https://alum.mit.edu/www/toms/paper/flexrbs/}
{https://alum.mit.edu/www/toms/paper/flexrbs/}",
year = "2001"}
see also
Below is an example lister map of the famous Lac promoter
region. The strong CRP site is shown along with the lac
promoter and the lacZ ribosome binding site. See:
https://alum.mit.edu/www/toms/paper/walker/
to understand sequence walkers and
https://alum.mit.edu/www/toms/paper/flexrbs/
for the method of making multi-part sequence walkers.
 A beautiful example is the cluster of Fis sites (in pink and
red shades) that control the Fis gene itself:
https://alum.mit.edu/www/toms/ftp/fispromoter.pdf
This is described in detail at:
https://alum.mit.edu/www/toms/fispromoter.html
A beautiful example is the cluster of Fis sites (in pink and
red shades) that control the Fis gene itself:
https://alum.mit.edu/www/toms/ftp/fispromoter.pdf
This is described in detail at:
https://alum.mit.edu/www/toms/fispromoter.html
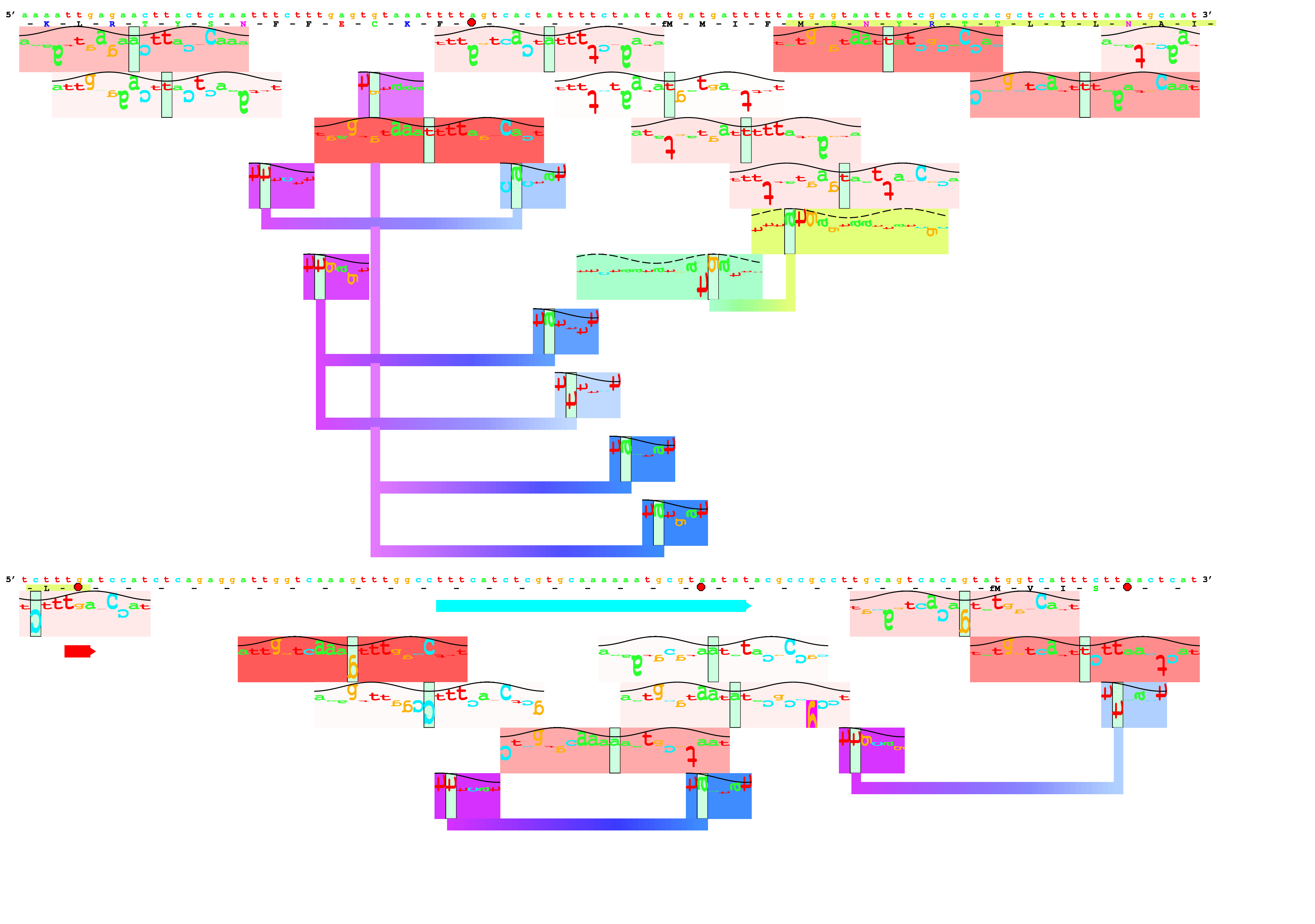 Example parameter file: listerp
Example features: lister.features
Example Delila instruction: lister.inst
Example petalfeatures file: petalfeatures
These were used to generate the map given above.
Example boundaries file: boundaries
Definition and examples of lister maps:
https://alum.mit.edu/www/toms/glossary.html#lister_map
Definition of lister features:
https://alum.mit.edu/www/toms/glossary.html#lister_feature
Definition of lister marks:
https://alum.mit.edu/www/toms/glossary.html#lister_mark
There are also examples in the Delila Tutorial:
https://alum.mit.edu/www/toms/delilainstructions.html
Related Programs, in approximate order of use:
delila.p, alist.p, split.p, dbmutate.p deprecated,
scan.p, makewalker.p, ri.p,
live.p, exon.p,
There are two versions of lister:
lister.p creates sequence walkers and is available by CDA, MTA or license:
https://alum.mit.edu/www/toms/contacts.html
listerx.p cannot create walkers and is available at this web site.
No longer generated.
For merging multiple marks files: mergemarks.p
Script: mkbookfeatures
Script to automatically create petals (colored rectangles)
behindcolor the sequence walkers: mkpetals
Script to put a time stamp on the map: listertimestamp
Top 10 things likely to be overheard if you had a Klingon Programmer:
http://rucus.ru.ac.za/~urban/docs/humour/Klingon%20Programmers.txt
author
Thomas D. Schneider
bugs
2004 Dec 5: the following line caused a crash of the
GPC compiler:
with f^, f^.definition^ do begin {causes GPC crash}
The next line should solve it:
with f^ do with f^.definition^ do begin
It would be possible to modify lister so as to report the amino acid
sequence in the features, but this raises some unresolved issues. The
position in the feature is a specific DNA base, so one could report the
amino acid in the reading frame that starts with that base. But then the
very next base will be in a different reading frame. Perhaps the solution
is to use the reading frame of the first base marked by the feature?
The parameter shiftsequence applies to every sequence, there is no
individual control.
"UPSIDE DOWN WALKERS"
Certain combinations of Delila instructions and searches can produce
incorrect walker listings. For example, if a binding site is even (ie
there is no central base) then to get the binding sites with Delila one
would say:
organism ecoli; chromosome ecoli; piece ecotrp;
get from 105 -20 to 105 +20;
get from 106 +20 to 106 -20;
(These are instructions to get the trpR sites from the siteli database.)
If these two same pieces are then scanned, scan will locate one
site on each of the pieces. There will, therefore, be two sites
listed in the data, scaninst and scanfeatures files:
@ ecotrp 105.0 +1 "TrpR" "@ 105|Ri=22.4|Z= 1.4210|p=0.078" 22.430327 1.421045 0.077652
@ ecotrp 106.0 +1 "TrpR" "@ 106|Ri=22.4|Z= 1.4210|p=0.078" 22.430327 1.421045
Lister will read the scanfeatures and apply them to EVERY piece in the
book. Thus it will apply both features to the first piece and both
features to the second piece. However, the results on the second piece
will be incorrect because it is the complementary sequence.
The walkers generally show up as highly negative.
A solution to this complex problem is not known. A practical solution is
to avoid scaning a sequences in both directions. That is, for the scan
only use one Delila instruction.
Should the walker really define the line to be it's own thickness? This
prevents one from putting character type features at any line except the
walker zero line.
No, it should not. Figure out how to get around this.
2001 Jan 4. PDF Font Problem. Before version 9.76 there was a problem
with conversion from PostScript to pdf by the gs (ghostscript) program.
Spaces were being compressed so that ascii features were shifted to the
left. This was solved by introducing a routine that puts multiple blanks
out (MB).
2001 Jan 5. However this then revealed a much worse problem which is that
the supposedly constant spacing courier font is not constant spacing for
parenthesis and dashes! This cannot be solved in lister directly. The
ultimate solution may be to rebuild lister so that every character is
placed onto the display at a particular coordinate, thereby getting around
the font failure AND allowing any kind of font to be used.
Because the symbols '(-)' are used for structures, special code was
created to solve this 'font compression' problem.
technical notes
The size of the page in PostScript is determined with 4 constants llx,
lly, urx and ury. These must be set correctly for each printer. These
can be easily obtained by printing the file:
https://alum.mit.edu/www/toms/ftp/printerarea.ps
In this program these parameters are set as 4 constants defaultllx,
defaultlly, defaulturx and defaultury. The parameters for pageedges
define whether to use the defaults or to compute the size to show.
The prgmod program defines the Ribl reading routines, in module scan.type
and getriblmatrix.
The featureinlist routine attempts to detect duplicate features. This can
happen when one obtains both the sequence and its complement for a site
and then one scans that book with the resulting model. How it is handled
depends on the symmetry. Odd and asymmetric cases are ok, but the even
case is difficult. It is detected by the zero bases being one position
apart with opposite orientations, but this will fail if the sites happen
to fall at a coordinate boundary. Though rare, at least the only result
would be an extra walker.
2000 Jun 28: 9.60: user definitions go into preamble!
The user may define in the marks file their own postscript code with "u",
and call it with "U". In the previous versions of the program the marks
were written 'on the fly', so that the file was read only once. This was
efficient, but caused a subtle bug. The color wave produced by the live
program would cause a problem. In ghostview, if one paged forward or
backwards quickly and the definition of the wave would be lost and
ghostview would freeze. This happend because the marks were put after the
postscript preamble, so they were not available for every page. The
solution was very simple (see usermarkstopreamble): put the user
definitions into the preamble. Then every page gets the defintions from
the preamble and ghostview can page very quickly. This also allows live
waves over multiple pages to be sent to our finicky (but correct) color
printer. Note that the user definitions are DUPLICATED in the parts after
the preamble. This means that the user can change definitions of graphics
throughout the map, which might be quite useful!
2006 Oct 20: /FontBBox is an array according to the PostScript
Language Reference Manual Second Edition (Red book):
/FontBBox: index on page 754
page 266-267: CLEARLY says it is an array!
font metrics and page 273
Ghostview does give an array, and all is perfect. However, the
COLOR 148 printer HP5550PS printer only returns three values ON THE
STACK instead of 4 values. I could not figure out what the three
values are. So in the letterrectangle routine (which is
responsible for making the color bar between multipart sequence
walkers) I used the ghostview values instead! This works but
obviously it may not work properly on all printers.
Bottom line: The HP printer appears to give an incorrect
result for /FontBBox.
google:
HP printer FontBBox
gives
http://www.ghostscript.com/doc/current/History2.htm
History of Ghostscript versions 2.n
"- buildfont required the presence of a valid, 4-element FontBBox.
(The Red Books say it's required, but Adobe interpreters don't
require it; some DEC software generates a 3-element FontBBox.)"
So it's true that there are 3-element FontBBoxes!
http://welcome.hp.com/country/us/en/contact/web_feedback.html
We have a Hewlett Packard LaserJet 5550 Printer. I am writing
postscript code that uses FontBBox. Although the PostScript
Language Reference Manual Second Edition (Red book) clearly states
on page 266-267 that this should return an array, your printer
returns three integers. That is a bug in your printer as far as I
can tell. I could not figure out what the values are. So: (1)
Please send this message to engineers who can deal with the bug in
your printer. (2) Please have them tell me what the three values
are. I need the standard llx, lly, urx and ury values of the
characters. How do I obtain these? Thank you.
Dr. Thomas D. Schneider
From: toms@alum.mit.edu
Sent: 2006-10-20 05:33:10Z (GMT)
Subject: HP U.S. Feedback: http://welcome.hp.com/country/us/en/prodserv.html
2006 Oct 26: 11.71: bug: acrobat 7.0 display on the sun is overwriting
the background under the walkers as one advances pages! The
background (normally white) becomes a colorful total mess. Change
routine startpage to have gsave as first thing. The display is still
ok, but the bug is still there. AH! page 13 was 'blank'. But when I
delete that page (by hand) it messes up anyway starting on page 14.
Acrobat display on the Mac is fine, this is a Sun-specific problem.
Deleting the first 9 pages in the ps problem gone from 10 until page
22 - ie actual page 13 again! Deleting to page 19, so it starts on
numbered page 20 - now the bug appears on page actual page 13 again!
So it's not the actual code, it's something about the length of the
material. Maybe some limit of Acrobat is being exceeded. Cannot
solve.
2012 Apr 18 for human sequences the numbers can be quite large. They
can bump into the astrisks: *138623230*. This is now ambiguous, so I
changed the '*' for 5 steps to be ',' giving: '*138623230,'.
2016 Aug 19: I switched functions to avoid using external coordinates
because a two-part walker over a circular coordinate bound cannot be
read in properly. Instead, each group is marked by 'group' and each
feature has a coordinate distance from the total zero.
gggg
****************************************************************************
Google:
"Top 10 things likely to be overheard if you had a Klingon Programmer"
http://rucus.ru.ac.za/~urban/docs/humour/Klingon%20Programmers.txt
Top 10 things likely to be overheard if you had a Klingon Programmer:
10) Specifications are for the weak and timid!
9) You question the worthiness of my code? I should kill you where you stand!
8) Indentation?! - I will show you how to indent when I indent your skull!
7) What is this talk of 'release'? Klingons do not make software 'releases'.
Our software 'escapes' leaving a bloody trail of designers and quality
assurance people in its wake.
6) Klingon function calls do not have 'parameters' - they have 'arguments'
- and they ALWAYS WIN THEM.
5) Debugging? Klingons do not debug. Our software does not coddle the weak.
4) A TRUE Klingon Warrior does not comment his code!
3) Klingon software does NOT have BUGS. It has FEATURES, and those features
are too sophisticated for a Romulan pig like you to understand.
2) You cannot truly appreciate Dilbert unless you've read it in the
original Klingon.
1) Our users will know fear and cower before our software! Ship it! Ship
it and let them flee like the dogs they are!
*)
(* end module describe.lister *)
{This manual page was created by makman 1.45}
Example parameter file: listerp
Example features: lister.features
Example Delila instruction: lister.inst
Example petalfeatures file: petalfeatures
These were used to generate the map given above.
Example boundaries file: boundaries
Definition and examples of lister maps:
https://alum.mit.edu/www/toms/glossary.html#lister_map
Definition of lister features:
https://alum.mit.edu/www/toms/glossary.html#lister_feature
Definition of lister marks:
https://alum.mit.edu/www/toms/glossary.html#lister_mark
There are also examples in the Delila Tutorial:
https://alum.mit.edu/www/toms/delilainstructions.html
Related Programs, in approximate order of use:
delila.p, alist.p, split.p, dbmutate.p deprecated,
scan.p, makewalker.p, ri.p,
live.p, exon.p,
There are two versions of lister:
lister.p creates sequence walkers and is available by CDA, MTA or license:
https://alum.mit.edu/www/toms/contacts.html
listerx.p cannot create walkers and is available at this web site.
No longer generated.
For merging multiple marks files: mergemarks.p
Script: mkbookfeatures
Script to automatically create petals (colored rectangles)
behindcolor the sequence walkers: mkpetals
Script to put a time stamp on the map: listertimestamp
Top 10 things likely to be overheard if you had a Klingon Programmer:
http://rucus.ru.ac.za/~urban/docs/humour/Klingon%20Programmers.txt
author
Thomas D. Schneider
bugs
2004 Dec 5: the following line caused a crash of the
GPC compiler:
with f^, f^.definition^ do begin {causes GPC crash}
The next line should solve it:
with f^ do with f^.definition^ do begin
It would be possible to modify lister so as to report the amino acid
sequence in the features, but this raises some unresolved issues. The
position in the feature is a specific DNA base, so one could report the
amino acid in the reading frame that starts with that base. But then the
very next base will be in a different reading frame. Perhaps the solution
is to use the reading frame of the first base marked by the feature?
The parameter shiftsequence applies to every sequence, there is no
individual control.
"UPSIDE DOWN WALKERS"
Certain combinations of Delila instructions and searches can produce
incorrect walker listings. For example, if a binding site is even (ie
there is no central base) then to get the binding sites with Delila one
would say:
organism ecoli; chromosome ecoli; piece ecotrp;
get from 105 -20 to 105 +20;
get from 106 +20 to 106 -20;
(These are instructions to get the trpR sites from the siteli database.)
If these two same pieces are then scanned, scan will locate one
site on each of the pieces. There will, therefore, be two sites
listed in the data, scaninst and scanfeatures files:
@ ecotrp 105.0 +1 "TrpR" "@ 105|Ri=22.4|Z= 1.4210|p=0.078" 22.430327 1.421045 0.077652
@ ecotrp 106.0 +1 "TrpR" "@ 106|Ri=22.4|Z= 1.4210|p=0.078" 22.430327 1.421045
Lister will read the scanfeatures and apply them to EVERY piece in the
book. Thus it will apply both features to the first piece and both
features to the second piece. However, the results on the second piece
will be incorrect because it is the complementary sequence.
The walkers generally show up as highly negative.
A solution to this complex problem is not known. A practical solution is
to avoid scaning a sequences in both directions. That is, for the scan
only use one Delila instruction.
Should the walker really define the line to be it's own thickness? This
prevents one from putting character type features at any line except the
walker zero line.
No, it should not. Figure out how to get around this.
2001 Jan 4. PDF Font Problem. Before version 9.76 there was a problem
with conversion from PostScript to pdf by the gs (ghostscript) program.
Spaces were being compressed so that ascii features were shifted to the
left. This was solved by introducing a routine that puts multiple blanks
out (MB).
2001 Jan 5. However this then revealed a much worse problem which is that
the supposedly constant spacing courier font is not constant spacing for
parenthesis and dashes! This cannot be solved in lister directly. The
ultimate solution may be to rebuild lister so that every character is
placed onto the display at a particular coordinate, thereby getting around
the font failure AND allowing any kind of font to be used.
Because the symbols '(-)' are used for structures, special code was
created to solve this 'font compression' problem.
technical notes
The size of the page in PostScript is determined with 4 constants llx,
lly, urx and ury. These must be set correctly for each printer. These
can be easily obtained by printing the file:
https://alum.mit.edu/www/toms/ftp/printerarea.ps
In this program these parameters are set as 4 constants defaultllx,
defaultlly, defaulturx and defaultury. The parameters for pageedges
define whether to use the defaults or to compute the size to show.
The prgmod program defines the Ribl reading routines, in module scan.type
and getriblmatrix.
The featureinlist routine attempts to detect duplicate features. This can
happen when one obtains both the sequence and its complement for a site
and then one scans that book with the resulting model. How it is handled
depends on the symmetry. Odd and asymmetric cases are ok, but the even
case is difficult. It is detected by the zero bases being one position
apart with opposite orientations, but this will fail if the sites happen
to fall at a coordinate boundary. Though rare, at least the only result
would be an extra walker.
2000 Jun 28: 9.60: user definitions go into preamble!
The user may define in the marks file their own postscript code with "u",
and call it with "U". In the previous versions of the program the marks
were written 'on the fly', so that the file was read only once. This was
efficient, but caused a subtle bug. The color wave produced by the live
program would cause a problem. In ghostview, if one paged forward or
backwards quickly and the definition of the wave would be lost and
ghostview would freeze. This happend because the marks were put after the
postscript preamble, so they were not available for every page. The
solution was very simple (see usermarkstopreamble): put the user
definitions into the preamble. Then every page gets the defintions from
the preamble and ghostview can page very quickly. This also allows live
waves over multiple pages to be sent to our finicky (but correct) color
printer. Note that the user definitions are DUPLICATED in the parts after
the preamble. This means that the user can change definitions of graphics
throughout the map, which might be quite useful!
2006 Oct 20: /FontBBox is an array according to the PostScript
Language Reference Manual Second Edition (Red book):
/FontBBox: index on page 754
page 266-267: CLEARLY says it is an array!
font metrics and page 273
Ghostview does give an array, and all is perfect. However, the
COLOR 148 printer HP5550PS printer only returns three values ON THE
STACK instead of 4 values. I could not figure out what the three
values are. So in the letterrectangle routine (which is
responsible for making the color bar between multipart sequence
walkers) I used the ghostview values instead! This works but
obviously it may not work properly on all printers.
Bottom line: The HP printer appears to give an incorrect
result for /FontBBox.
google:
HP printer FontBBox
gives
http://www.ghostscript.com/doc/current/History2.htm
History of Ghostscript versions 2.n
"- buildfont required the presence of a valid, 4-element FontBBox.
(The Red Books say it's required, but Adobe interpreters don't
require it; some DEC software generates a 3-element FontBBox.)"
So it's true that there are 3-element FontBBoxes!
http://welcome.hp.com/country/us/en/contact/web_feedback.html
We have a Hewlett Packard LaserJet 5550 Printer. I am writing
postscript code that uses FontBBox. Although the PostScript
Language Reference Manual Second Edition (Red book) clearly states
on page 266-267 that this should return an array, your printer
returns three integers. That is a bug in your printer as far as I
can tell. I could not figure out what the values are. So: (1)
Please send this message to engineers who can deal with the bug in
your printer. (2) Please have them tell me what the three values
are. I need the standard llx, lly, urx and ury values of the
characters. How do I obtain these? Thank you.
Dr. Thomas D. Schneider
From: toms@alum.mit.edu
Sent: 2006-10-20 05:33:10Z (GMT)
Subject: HP U.S. Feedback: http://welcome.hp.com/country/us/en/prodserv.html
2006 Oct 26: 11.71: bug: acrobat 7.0 display on the sun is overwriting
the background under the walkers as one advances pages! The
background (normally white) becomes a colorful total mess. Change
routine startpage to have gsave as first thing. The display is still
ok, but the bug is still there. AH! page 13 was 'blank'. But when I
delete that page (by hand) it messes up anyway starting on page 14.
Acrobat display on the Mac is fine, this is a Sun-specific problem.
Deleting the first 9 pages in the ps problem gone from 10 until page
22 - ie actual page 13 again! Deleting to page 19, so it starts on
numbered page 20 - now the bug appears on page actual page 13 again!
So it's not the actual code, it's something about the length of the
material. Maybe some limit of Acrobat is being exceeded. Cannot
solve.
2012 Apr 18 for human sequences the numbers can be quite large. They
can bump into the astrisks: *138623230*. This is now ambiguous, so I
changed the '*' for 5 steps to be ',' giving: '*138623230,'.
2016 Aug 19: I switched functions to avoid using external coordinates
because a two-part walker over a circular coordinate bound cannot be
read in properly. Instead, each group is marked by 'group' and each
feature has a coordinate distance from the total zero.
gggg
****************************************************************************
Google:
"Top 10 things likely to be overheard if you had a Klingon Programmer"
http://rucus.ru.ac.za/~urban/docs/humour/Klingon%20Programmers.txt
Top 10 things likely to be overheard if you had a Klingon Programmer:
10) Specifications are for the weak and timid!
9) You question the worthiness of my code? I should kill you where you stand!
8) Indentation?! - I will show you how to indent when I indent your skull!
7) What is this talk of 'release'? Klingons do not make software 'releases'.
Our software 'escapes' leaving a bloody trail of designers and quality
assurance people in its wake.
6) Klingon function calls do not have 'parameters' - they have 'arguments'
- and they ALWAYS WIN THEM.
5) Debugging? Klingons do not debug. Our software does not coddle the weak.
4) A TRUE Klingon Warrior does not comment his code!
3) Klingon software does NOT have BUGS. It has FEATURES, and those features
are too sophisticated for a Romulan pig like you to understand.
2) You cannot truly appreciate Dilbert unless you've read it in the
original Klingon.
1) Our users will know fear and cower before our software! Ship it! Ship
it and let them flee like the dogs they are!
*)
(* end module describe.lister *)
{This manual page was created by makman 1.45}
{