 ev: Evolution of Biological Information,
ev: Evolution of Biological Information,
In approximate decreasing order of importance:
Method to find window size relative to Terminal size:New as of 2014 Oct 21.
Use Terminal and match it to a Evj window while running this script:while (1) echo;echo -n 'lines: '; tput lines ; echo -n 'columns:' ; tput columns; sleep 1 end--- RESULT:
lines: 46 columns:104
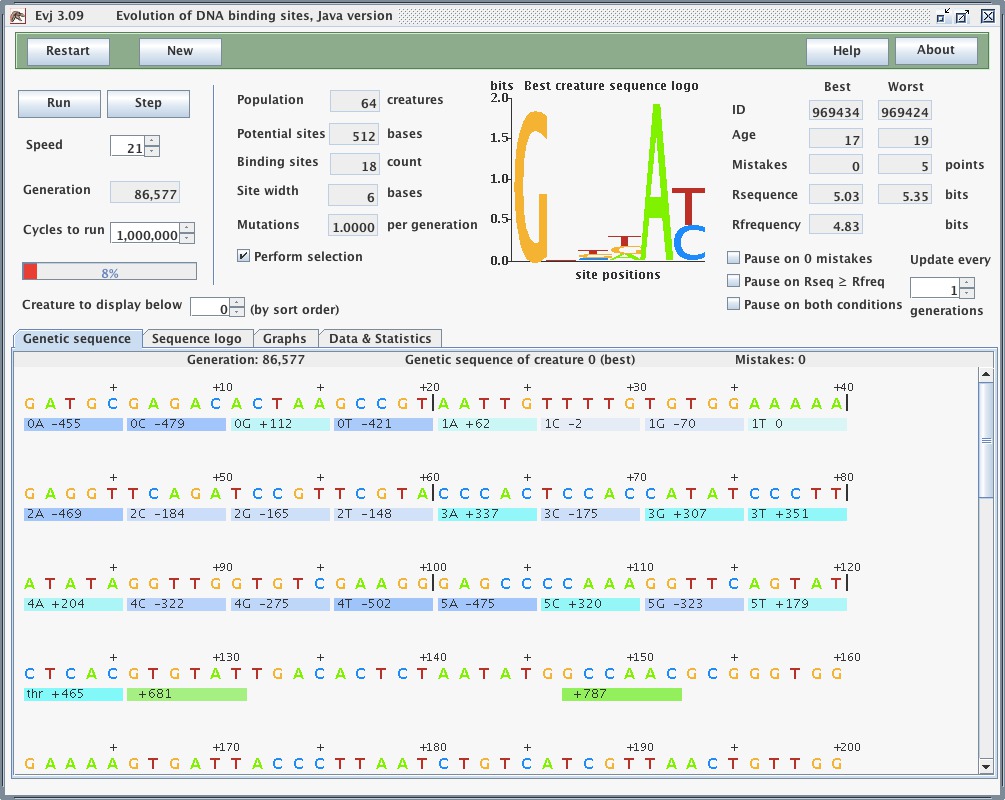
Logo of whole genome as an option! One would specify the number of previous generations to include. The program would have to capture the frequency data for the whole genome for that many generations, which could be expensive in memory. To change the data every generation, one would need to keep every previous generation (over the desired time interval) so that the bases from the last genome could be removed.
Logo of whole genome: time slice This would show a logo of all organism genomes in the current time slice.
Statistics
PA:
A way to avoid this intense memory use would be to restart the frequency data at zero every so many generations. Once that many generations have passed, display the logo. Then zero and gather for the next. So this would require two genome-length arrays, the current one being displayed and the one being built.
Paul: Support for variable mistake points is included.
Tom: Oh my!!! I set missed site to be 10 and the other to be 1 and it evolved in only 316 generations instead of 700 or so! (actual number: 662)
Paul: Yes, isn't that cool?
Tom: Very. It makes me think of a higher level of programming. I'm not sure how this would work but suppose that I wanted to try a range of variable mistake points. Doing this by hand would be tricky. The old Ev in Pascal lets one call the thing in a loop, as I did a few days ago to answer a question you raised.
So suppose that the user could request a range of values of some parameter. The user would specify number of repeats using different values of the random number generator. So the super controller would run through that parameter, repeating each several times. For example, the time to the first perfect creature would be on the y axis and the site weight on the x axis. This would let someone do a real experiment and plot the result automatically.
This might be a pretty big change from where things are now, but it would allow more questions to be answered easily.
crossingcounter := 0;
crossinglimit := (from user);
AboveRf := false;
compute Rf;
/* start the main loop */
while (otherlimits) and (crossingcounter <= crossinglimit) {
(compute Rs)
if ((Rf > Rs) and (not AboveRf))
or
((Rf < Rs) and ( AboveRf))
then {
crossingcounter := crossingcounter + 1;
AboveRf := not AboveRf
}
}
2006 Jun 11: Factors not in Evj, Tom and Paul:
2006 Jun 11: Evj control program. Tom: A control program could be built that sets parameters instead of a person. It would allow experiments to be done more easily by stepping a defined loop that controlled variables. It is possible with the original Ev, but it's hard with the Evj interface. The output should be at minimum cut and pastable, better would be to be able to write it to a file for further analysis. An alternative is to allow calling Evj from the command line but then most people would not do the experiments.
2006 Jun 19: Tom: Provide mechanism to keep the 'new' panel up all the time. This should make experimentation easier since the user wouldn't have to keep bringing it up all time.
2006 Jun 20:
Display message for refusal to accept a New parameter
Alan Klein:
I click the "ok" button on the form to create a new model. The performance
icon from the task manager goes to 100% for about 1 second then goes back to
it's base cpu usage level and the form doesn't close. All the other entry
fields and buttons work but it won't accept the large population values. If
I reduce the population down, the "ok" button works again, the form closes
and you can run those values in ev. No error message is displayed and the
mouse cursor stays as an arrow. If the program is doing internal processing
such as loading data into memory or using a disk swap file, perhaps you
could change the mouse pointer to the hour glass.
Tom: Confirmed. In New, if you set very large population values, (eg 100,000 creatures) then when one clicks OK, the cpu maxes out (as observed with top under unix for example) and the New panel does not go away.
Paul: I believe if you watch your Java console, you will see:
Java.lang.OutOfMemoryError: Java heap spaceI may be able to catch that error and display a more reasonable message.
2018 May 21:
Switch for two's complement to binary evaluation
Ev and evj use two's complement notation for the evaluation.
This should not be essential, so a switch to binary (with a higher
threshold) or other methods (what?) should still give the
same evolutionary results.
People seem to have a hard time visualizing the horrific number of deaths that occur during an Ev run. Maybe a counter 'Number of deaths' under the logo would emphasize it? It would be reset to zero when things are restarted.
Obviously 50% of the population dies every generation. If there are 64 creatures and lesee ... 675 generations to Rs >= Rf :-) :-) a reproducible result! :-) then 32*675=21600 creatures died to get 4 bits of information per site - with 16 sites that is 64 bits or MORE (you've seen 68%): roughly 337 deaths per bit. Given that each selection removes half of the population, it COULD HAVE done 337 bits. So it is extremely inefficient, 1/337 = 0.3%.
Another statistic that might be more enlightening would only count deaths IF mistakes is > 0. That is, once the system stabilizes with Rs~Rf, many deaths are just luck of the draw. The creature happened to be below the line but had no mistakes. Tough luck! But if mistakes > 0 the creature can die because it is selected against.
So we could have:
deaths deaths by mistakeThe problem when people look at a beautiful living thing like a tree is that they don't notice the stunted ones near by, the seeds that didn't germinate, the ones that crashed over in a storm. Deaths are often invisible. By putting the number on the display, it would not be overlooked.
Paul: How about counting deaths if the number of mistakes is greater than the worst creature left alive? Then we don't count deaths of creatures who are as good as the ones left alive, but who are dying just by accident.
Tom: That's not quite right - the worst creature isn't the critical one because that one is not the reason for THIS creature's death.
Let's see. First, the creature is in the half that dies. The question is: are there any creatures better than that who would be "responsible" for this creature dying?
So wouldn't the criterion be that there is at least one creature with a number of mistakes fewer than the one that is dying?
So - determine the mininmum number of mistakes (already done) and if a creature dies AND has more mistakes than this minimum, we count the death.
The 'horrific' thing will be to see how this keeps on climbing after Rs = Rf or mistakes = 0!
There can be a graph of death by mistake.
Paul: But the worst one left alive, the guy just above the midpoint of the sorted list, is no better than the best one killed, if their scores are equal. Which one of those two dies is just a crapshoot.
But why single out the one with the fewest mistakes? A guy 1/4 of the way down the sorted list is just as responsible for the deaths of the bottom half, but is closer in score.
This doesn't seem right.
Tom:
Ok, I see. Your suggestion to
count death because of having more
mistakes than the worst creature left alive
is good.
[done earlier than 2006 Jun 17 version 2.35]
We have the space but it is so important that I think an option for both should be another check box. Maybe the Rs ≥ Rf option alone or the counting option would go onto the New panel later. [done on 2006 Jun 17 version 2.35]
2006 Jun 17: Mutations per base.
Tom: Experiments should be done with constant mutations per base. It is UNREASONABLE to increase genome size and simultaneously effectively decrease the mutation rate per base because we know that mutation comes from polymerase copying error and from exposure to mutagens. Both of those are on a per base basis.
Paul: Good point. What sort of rate should we use? Hey, maybe Evj needs a mutations/base rather than just a fixed number.
Tom: Yes. The question is how to implement it without it costing a lot.
Tom: Let's see. Suppose we said 1/256 mutations per base. Then that would be 1 per genome that is 256 long. So we compute 256 * 1/256 ~ 1 and use 1 per genome. This would work for even multiples: 512 * 1/256 ~ 2 per genome. It would be inexpensive to set up. So what do we do if it's not an exact multiple?
Paul: Round to the nearest integer, I would think. Tom: That would be ok but not the best.
Tom: Hmm. One way is to do the integer part of the number per genome. Then flip ONE random number for the remaining part. Sure! That's one random number (expensive) per generation and creature. Not too bad compared to one random number per BASE copied! We have (say) 2.2 as our mutations per genome (computed from mutations per base). So we do 2 hits and then flip a random number between 0 and 1. If the random number is between 0 and 0.2, we do another hit, otherwise not. On the long run you get 2.2 hits per genome.
Paul: That sounds better. Do we specify mutations/base, or > mutations/kilobase so the user doesn't have to specify real numbers?
Tom: It would be easier on the person to give it as mutations/base. So there would be the original method (hits per genome) and a toggle to get the new methods (hits per base as a real number). Maybe the user would input one over hits per base, so an input of 256 means one hit every 256 bases or 1 hit per genome. NOTE that this method is not going to give the full proper poisson distribution. That's pretty expensive because it has to be done for every base.
Summary: Provide mutations per base as a real number: "1 in every ______ bases" Compute: m = genome size * mutations / bases. To create mutations in an organism, split m into integer (mi) and decimal (md) parts. Do the integer part of m mutations. Chose a random number r between 0 and 1. If r <= md, do an additional mutation. In this way the requested mutations will be done on average.
[2006 Jun 19] Paul: Regarding the two methods of determining mutations per genome: When we divide genome size by (1 mutation per) n (bases), do we want to use the potential sites count or the full size of the chromosome (remember, it's padded so a binding site can occur at the end).
Tom:
Tricky! Let's see, in the standard example it's 261 bases long with
256 potential sites.
Mutations need to go into the 261 bases.
But if we force the person to think about the
extra padding it will be a pain.
Tentatively let's say the user asks to use one mutation every 256
bases. Then we know the precise rate. That rate applies, of course,
to the padding at the end. So the computation would have to be with
261. Suppose that the person says that there should be one mutation
every 256 bases and the potential sites are 500 long. Then with 6
site width sites, that's 505. We compute with 505.
2006 Jun 21: Done in version 2.36.
![]()

Schneider Lab
origin: 2005 Aug 3
updated: 2020 May 21: google: can you run java on a cell phone iOS
![]()