1
START SLIDE SHOW Launch blank background browser. This should work on any browser but will not give full screen coverage.* Popup blank background browser. This gives full screen coverage but depends on javascript so may not work on all browsers. Click on the link to get this page to begin the slide show.*
 2
2
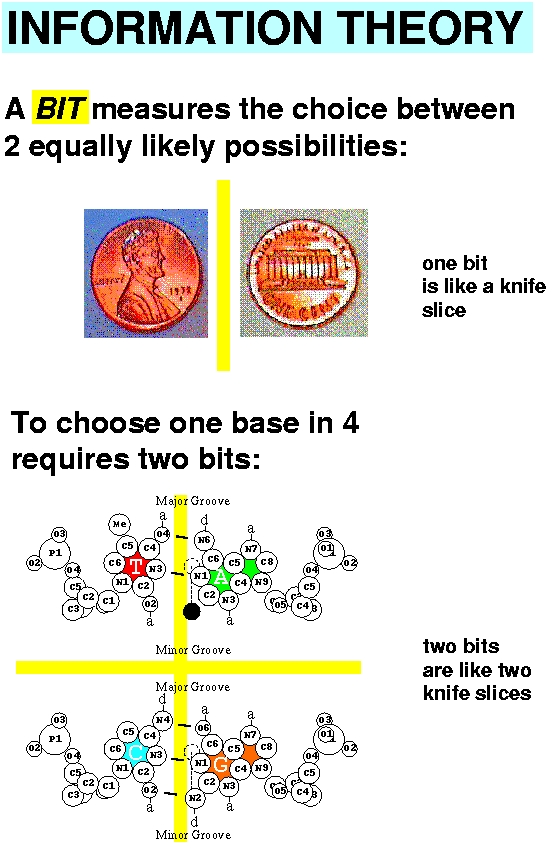 3
3
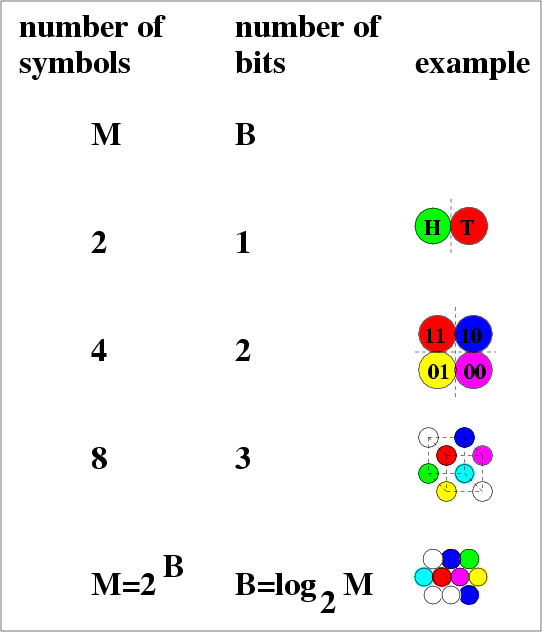 4
4
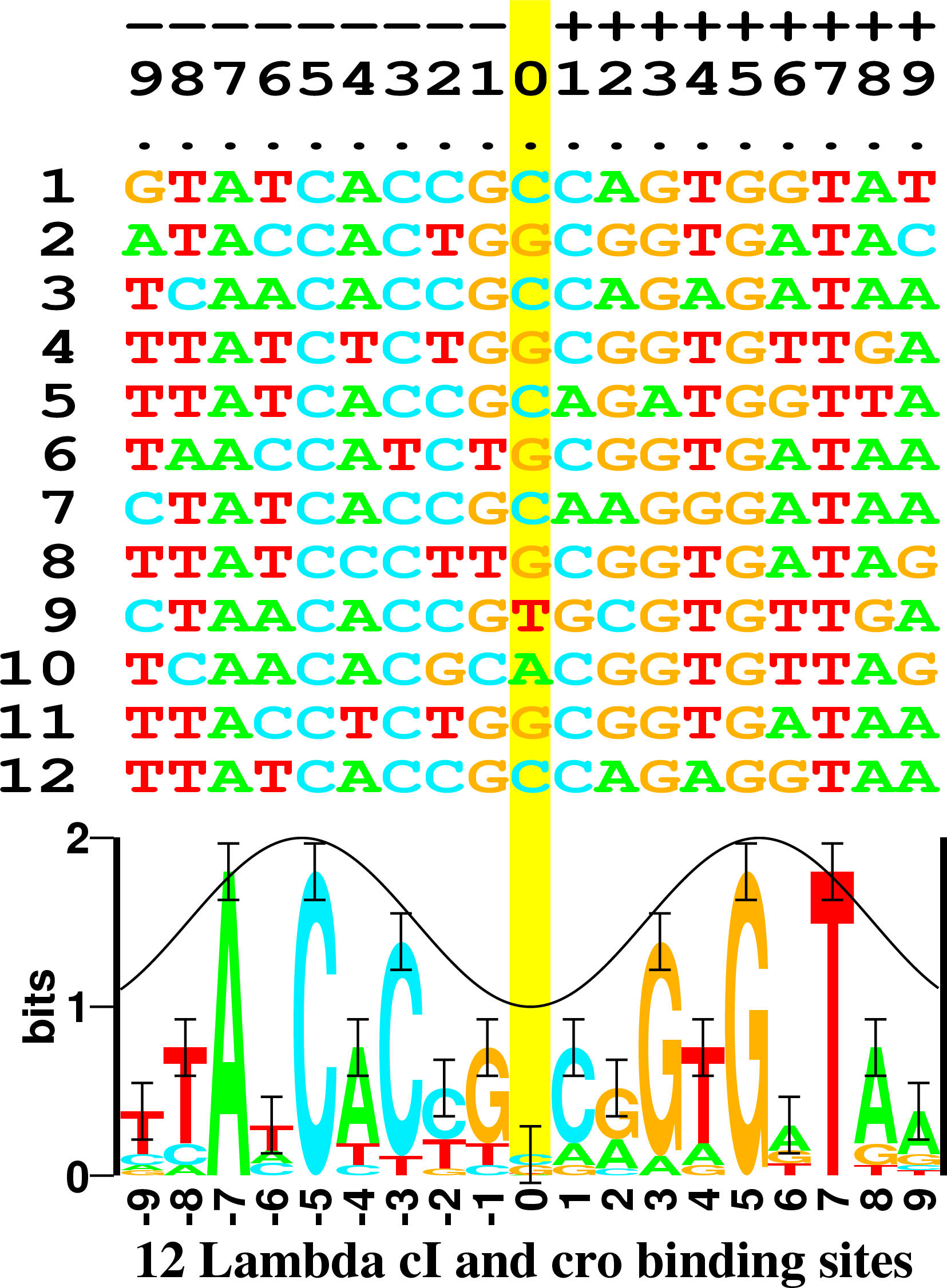 5
5
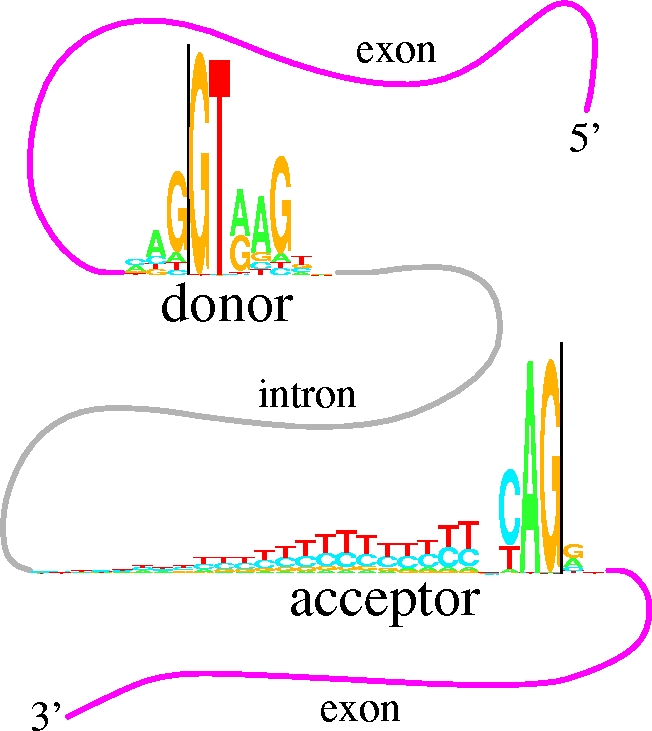 6
6
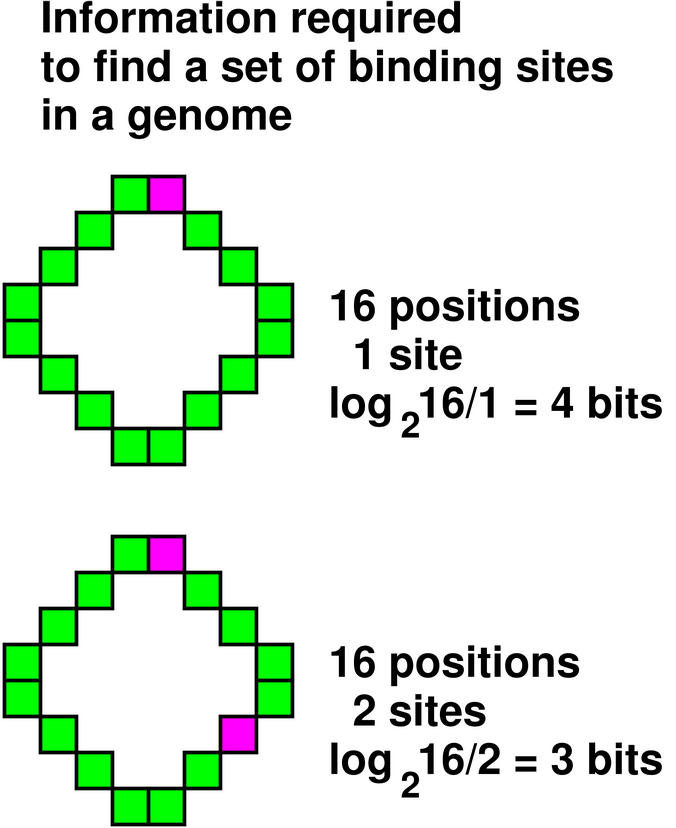 7
7
 8
8
 9
9
 10
10
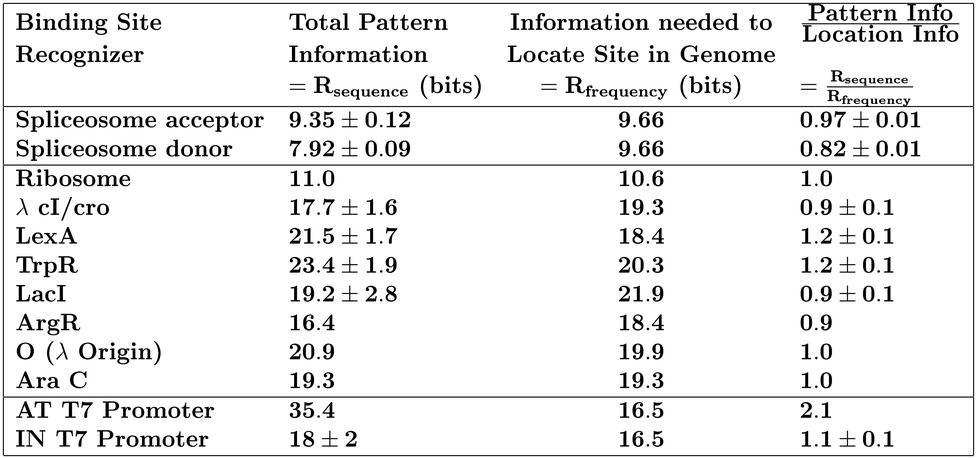 11
11
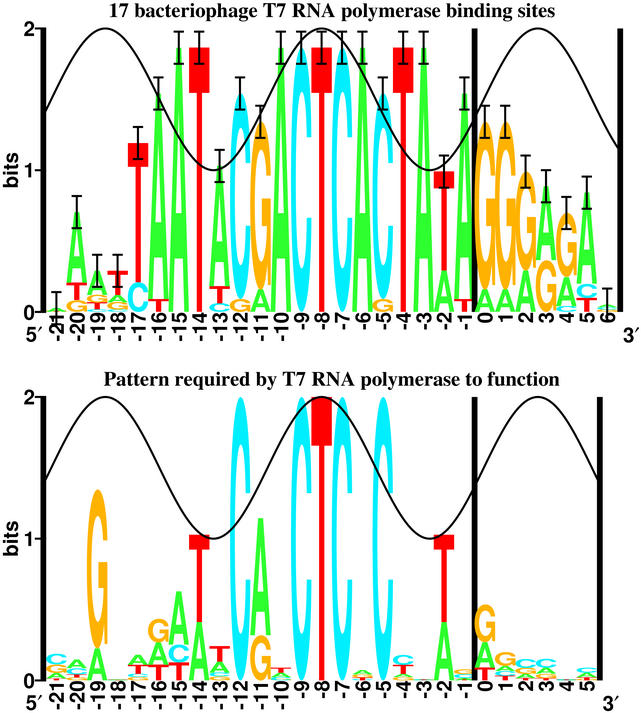 12
12
 13
13
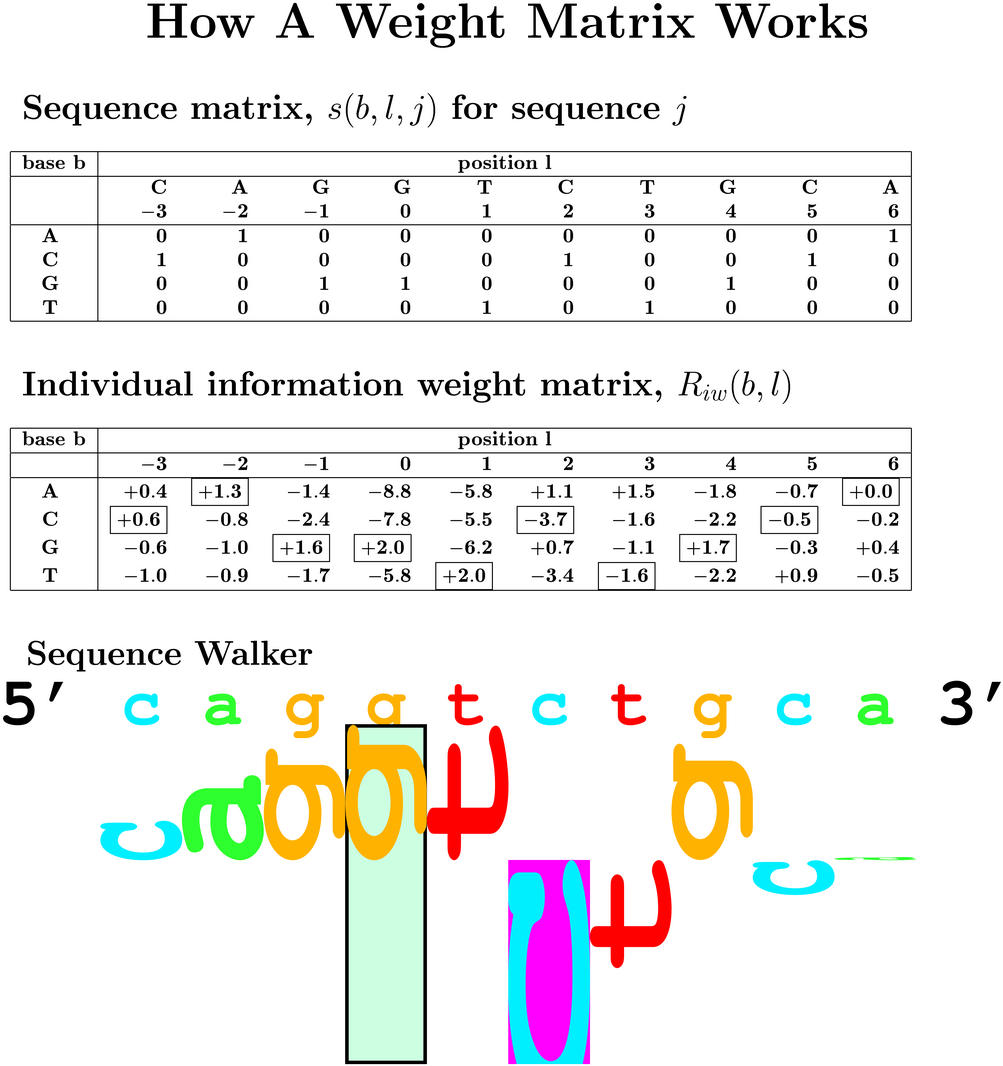 14
14
 15
15
 16
16
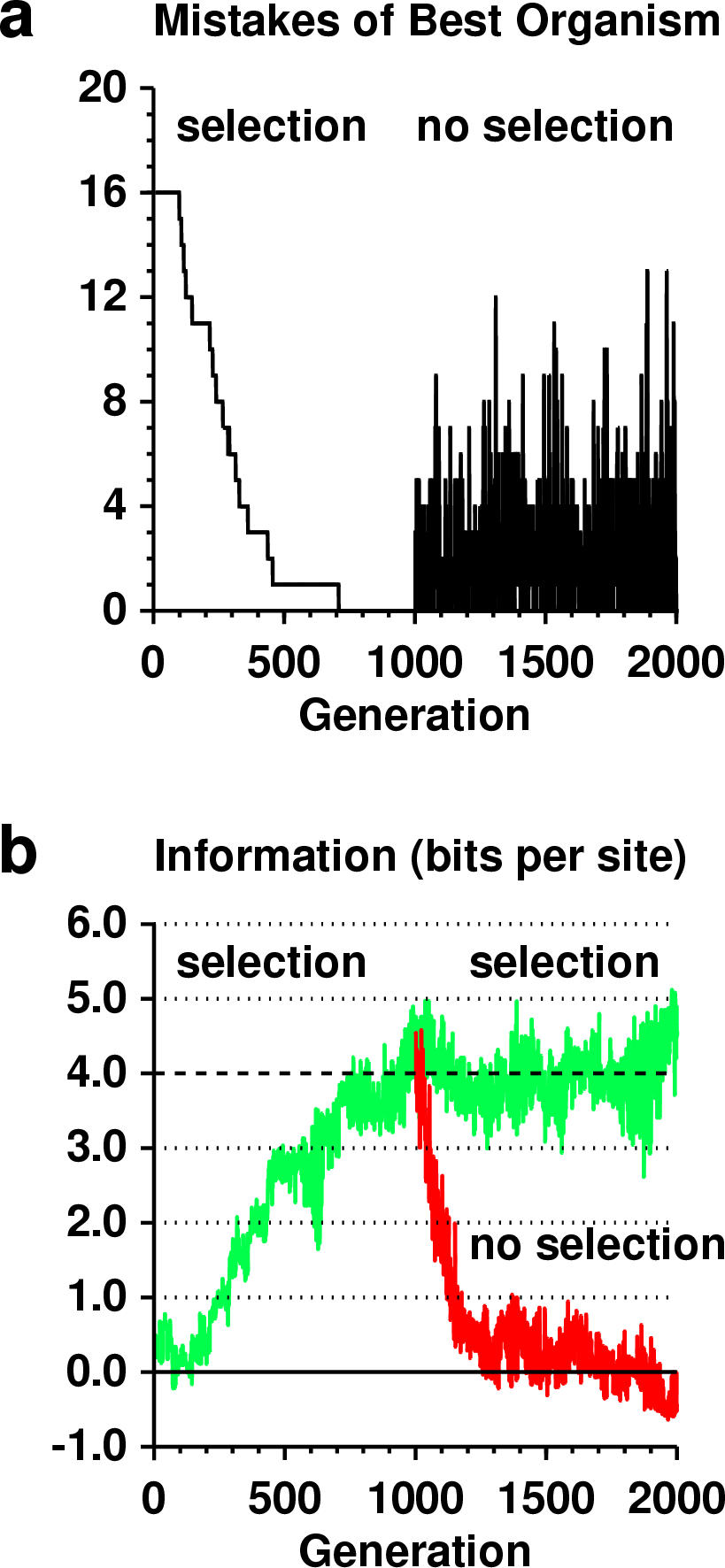 17
17
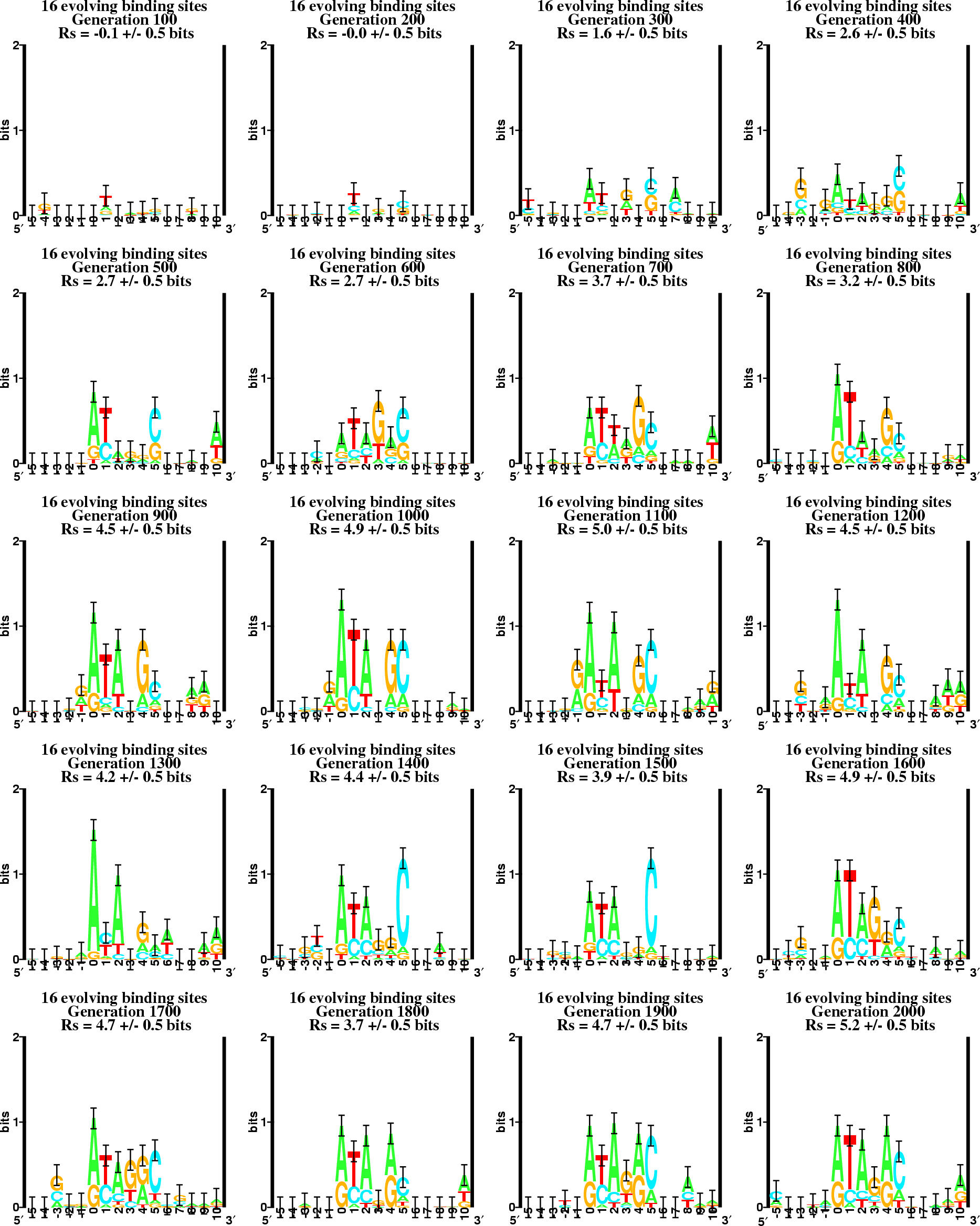 18
18
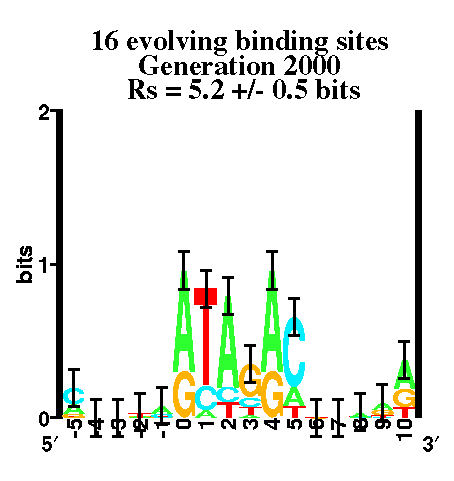 19
19
 20
20
 21
21

Printing options:
This slide show was created
on
2011Aug17.20:35:55
by the slideshow script:
version = 2.22 of slideshow 2011 Aug 17: make a slide show
https://alum.mit.edu/www/toms/ftp/slides
https://alum.mit.edu/www/toms/slideshow
author:
Tom Schneider