Instructions on How to Refine a Flexible Information-Theory Based
Ribosome Binding Model From an Initially Annotated Genome.
By Ryan Shultzaberger
version = 1.05 of refineinst.html 2005 Oct 13
Introduction:
These instructions were written to help scientists familiar with the programs
used in Dr. Thomas Schnieder's lab, to refine a flexible information-theory
based ribosome binding model from an initially annotated genome as done
previously
(Shultzaberger.Schneider2001). If you are unfamiliar with the programs
used in Dr. Schneider's lab you can learn more about them at: https://alum.mit.edu/www/toms/ .
Theory:
Rigid information-theory based models are not appropriate for the
modeling of flexible binding systems, like ribosomes. Flexible binding
systems are those systems that contain more than one binding element with
some variable distance between those elements. Prokaryotic ribosome
initiation relies on the 30S recognition of the correct start codon and a
rRNA-mRNA hybridization between the 16S rRNA and a region upstream of the
start codon (the Shine-Dalgarno). The spacing between the Shine-Dalgarno and
the start codon varies, making this system a flexible binding system. There
is an in-depth description of the mathematics used to characterized flexible
binding systems in
Shultzaberger.Schneider2001 .
The number of sequenced genomes is increasing rapidly. These genomes are
often annotated by computer algorithms that are fairly, but not completely,
accurate. These programs work by looking at reading frame size, G/C content
in the third position, and homology between genes in other organisms. These
initial annotations often have incorrectly annotated genes. The
methodology described in this document can be used to help correct these
misplaced starts.
A flexible binding model can be made of all starts in an initially annotated
genome using methods previously discussed (
Shultzaberger.Schneider2001) . The individual information scores of each site
within the set can be determined. These individual information scores
reflect the relative strength of each site within the total set. The
relative frequency of each nucleotide base at each position is determined,
for the whole set, creating a weight matrix. Then each site is compared to
the weight matrix, and the individual score for that site can be determined.
If the site has an individual information score greater than zero bits, then
this site is thought to be real. A positive information score corresponds
to a negative delta G. Conversely, if a site has a negative score (or a
positive delta G), then the site is not real and the ribosome will not bind
to it.
The refinement method, which is the method for which these instructions are
for, is a means to remove, identify, and eventually adjust these incorrect
sites. After the model is made and those sites with negative information are
identified, they are removed and the model is rebuilt. After this model is
rebuilt there is a re-evaluation of the individual scores of each site. New
sites that now have negative information, as compared to the new total set,
are then identified and removed. This process of rebuilding and removing
continues until no negative sites remain in the set. Once no more sites
remain, the model is said to be refined.
This refined model is assumed to be made of correct sites and can be further
used to predict where the correct initiation site is for those removed
presumably incorrectly annotated sites. Examples of this are also within
Shultzaberger.Schneider2001 .
Programs used:
wgetac
exon
instshift
catal
delila
alist
encode
rseq
dalvec
makelogo
ri
embed
malign
malin
diffinst
genhis
xyplo
diri
makeinst
Instructions:
A. Make a clean directory to work in (dir xxx), and
make the following directories within this new directory:
1. malignsd
2. rounds
3. discan
(Here is a diagram of the final directory structure .)
B. In directory xxx you will make your first model, which is
an aligned listing of gene starts from GenBank.
To do this do the following:
1. Use wgetac to get your selected genome off of GenBank.
This works by typing "wgetac ACCESSION_NUMBER".
2. Copy this file, which is named ACCESSION_NUMBER to db.
3. Run exon on db, but make sure that the "acceptor range is +/- 100"
in the exonp file.
4. Use instshift so that the first base in the p-site is the zero
position in your model.
5. Generate your lib and cat files that support your new genome.
6. Manipulate the ainst file so that it is in the following format:
a. "get from" is at the left of each line. This means, you need
to remove the piece and name from each line, but make sure to
keep the first piece so that delila will work.
b. You need to sort and order the inst file so that it is arranged
lowest to highest coordinate in the positive orientation, followed
by lowest to highest in the negative orientation.
Here is an example inst file: example inst
7. Using this inst file, make a logo using
run.logo (current version).
(run.logo version 1.18 was originally used.)
8. Copy this inst file to rounds/inst.atg.0 and to malignsd/inst .
C. With this inst file you can multiple align out your Shine-Dalgarno.
To malign the Shine-Dalgarno do the following in the malignsd directory:
1. Embed out the region associated with the P-site, so that it does not bias
your re-alignment (like in the figure below). Make sure to copy the
embedbk to book. (example embedp file)

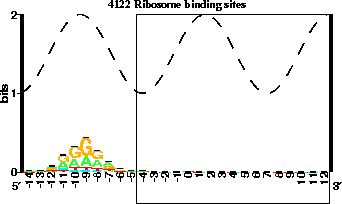
Figure 1: The region associated with the p-site is embedded out.
2. Run malign over the region of the SD, and look at the malignxyin output
to determine the best alignment.
(example malignp file)
3. Run malin to make an inst of the best one.
4. Copy this realigned inst to .. /rounds/inst.sd.0
5. Now go into the rounds directory and use instshift to shift the maligned
inst file so that the central base is at the zero position.
You now have two inst files (inst.atg.0 and inst.sd.0). Your
SD model should look like the following:

Figure 2: Your model should look like this, notice how the central position
is at base zero.
D. Go into your discan directory and make two new directories:
1. simul.rounds
2. diri
E. In simul.rounds/ you will generate your rixyin files to be used by diri.
Do the following:
1. Copy your inst.sd.0 file to inst in the simul.rounds directory, and to
insta in the simul.rounds directory.
2. Run delila, alist, encode, rseq, dalvec, makelogo, and ri to generate an
rixyin file for later use. Make sure that your rip file is for the
range that you want, for the sd region. Copy this file into;
.. /diri/bri .
3. Copy your inst.atg.0 file to inst in the simul.rounds directory, and to
instb in the simul.rounds directory.
4. Run delila, alist, encode, rseq, dalvec, makelogo, and ri to generate an
rixyin file for later use. Make sure that your rip file is for the
range that you want, for the initiation region. Copy this file into;
.. /diri/ari .
5. You need to generate a distribution of spaces between the SD and ATG models
to do this run diffinst and genhis on the insta and instb files you made
in steps 1 and 3. Copy the histog output .. /diri/histog .
F. Now that you have your rixyin files for both the SD and AUG part of the model,
as well as the distribution of the spacings, you can determine the individual
information contents of each site in the model. In order to determine the
individual information content, do the following:
1. In the diri directory, run diri and look at the dcout file.
G. Now you only want those sites in the dcout file that have positive
information.
1. The data input file for makeinst is composed of a coordinate and an
information score. Since you are only interested in those sites that are
positive, you can grep -v out the negative scores. Here is a possible script
that can be used to make this data file: possible script
2. Run makeinst on the new data file to generate new (the data.atg output
from the above script), refined inst files, to generate instructions for
these sites. Notice that these instructions do not have header material, you
need to insert this.
3. Copy this new inst to xxx/rounds/inst.atg.1 . This is the inst file for
the initiation region after the first round of cleaning.
H. Use this inst file like you used inst.atg.0, and repeat steps C-G,
skipping step D. Each time you repeat make sure to raise you increments by 1
so that after the second round you would have inst.atg.2 and inst.sd.2
Continue to repeat until you have no more negative sites in your model. This
is your refined model.


Schneider Lab
origin: 2002 Jan 22
updated: see top.