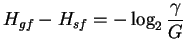If a genome contains G bases, there are M=G
ways that its sequence can be aligned or G
potential binding sites.
If these are all equally likely,
then Pi = 1/G and formula (1) reduces to:
If the genome contains ![]() sites,
we assume that the probabilities of binding to each
site are equal
and that the probability of significant binding to other sequences is
zero.
This allows formula (1) to be reduced to:
sites,
we assume that the probabilities of binding to each
site are equal
and that the probability of significant binding to other sequences is
zero.
This allows formula (1) to be reduced to:
The decrease in positional uncertainty during binding or
alignment is the difference:
Rfrequency is the amount of information needed to pick
![]() sites out of G possible sites.
As the number of sites in the genome increases,
the information needed to find a site
decreases.
As long as
the simplifying assumption for equation (8) holds and
sites out of G possible sites.
As the number of sites in the genome increases,
the information needed to find a site
decreases.
As long as
the simplifying assumption for equation (8) holds and ![]() is
restricted to the number of known sites (that is,
is
restricted to the number of known sites (that is, ![]() is not an estimate), equation (9)
gives an upper bound on
Rfrequency since some sites may exist that are not
now known. A second property of this formula is that
Rfrequency is
insensitive to small changes in G or
is not an estimate), equation (9)
gives an upper bound on
Rfrequency since some sites may exist that are not
now known. A second property of this formula is that
Rfrequency is
insensitive to small changes in G or ![]() .
The frequency of sites must
change by a factor of two to alter
Rfrequency by only one bit.
The largest
possible value of
Rfrequency occurs for a single site in the genome:
.
The frequency of sites must
change by a factor of two to alter
Rfrequency by only one bit.
The largest
possible value of
Rfrequency occurs for a single site in the genome:
![]() .
(For E. coli,
Rfrequency = 22.9 bits in this case.)
On the other hand, if all
positions in the genome were sites, one would not need any information to find
them, and
Rfrequency would be zero.
.
(For E. coli,
Rfrequency = 22.9 bits in this case.)
On the other hand, if all
positions in the genome were sites, one would not need any information to find
them, and
Rfrequency would be zero.
The number of potential binding sites (G) is twice the number of base
pairs in a DNA genome because there are two orientations for a recognizer to
bind at each base pair. A symmetrical recognizer on DNA has two ways to bind
each base pair, and both ways are used at a binding site.
Here, ![]() is twice the number of conventional binding sites. An asymmetric recognizer
on DNA will use only one orientation at any particular base pair. In this
case,
is twice the number of conventional binding sites. An asymmetric recognizer
on DNA will use only one orientation at any particular base pair. In this
case, ![]() is equal to the number of binding sites.
On RNA there is only one
possible orientation. Thus G and
is equal to the number of binding sites.
On RNA there is only one
possible orientation. Thus G and ![]() reflect not only the genome size and
number of binding sites but also the symmetries of the recognizer and nucleic
acid.
reflect not only the genome size and
number of binding sites but also the symmetries of the recognizer and nucleic
acid.