 Dissection of
"A Vivisection of the ev Computer Organism:
Identifying Sources of Active Information"
Dissection of
"A Vivisection of the ev Computer Organism:
Identifying Sources of Active Information"
This is a brief (and perhaps preliminary) response to:
@article{Montanez.Marks2010,
author = "G. {Monta\~{n}ez}
and W. Ewert
and W. Dembski
and R. Marks",
title = "{A Vivisection of the ev Computer Organism: Identifying
Sources of Active Information}",
journal = "BIO-Complexity",
volume = "2010",
pages = "1--6",
url = "http://bio-complexity.org/ojs/index.php/main/article/view/36"
year = "2010"}
The abstract of the above paper claims:
"ev is an evolutionary search algorithm proposed to simulate biological evolution. As such, researchers have claimed that it demonstrates that a blind, unguided search is able to generate new information."
That's not what is claimed in the original paper, Evolution of Biological Information.
There are two parts to evolution: replication with variation AND selection. In addition to mutational variation, the ev program has selection and so it is does not do an 'unguided search'.
 Chris Adami has pointed out that
the genetic information in biological systems comes from the environment.
In the case of Ev, the information
comes from the size of the genome (G)
and the number of sites (γ), as stated clearly in the paper.
From
G
and
γ
one computes
Rfrequency = log2 G / γ bits per site,
the information needed to locate the sites in the genome.
The information measured in the sites (Rsequence, bits per site)
starts at zero and
entirely by an evolutionary process
Rsequence
converges on
Rfrequency.
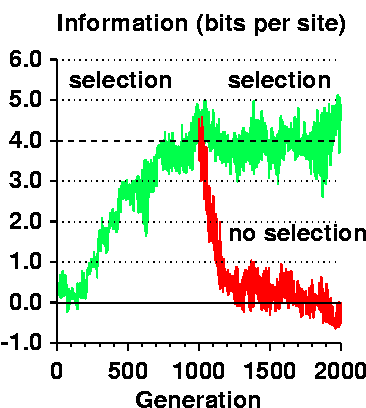
In the figure to the right,
Rfrequency is shown by the dashed line and
the evolving Rsequence is the green curve.
At 1000 generations the population was duplicated and in one
case selection continued (horizontal noisy green curve)
and in the other case selection was turned off
(exponentially decaying noisy red curve).
Thus the information gain depends on selection and it not blind and unguided.
The selection is based on biologically sensible criteria: having
functional DNA binding sites and not having extra ones. So Ev models
the natural situation.
Chris Adami has pointed out that
the genetic information in biological systems comes from the environment.
In the case of Ev, the information
comes from the size of the genome (G)
and the number of sites (γ), as stated clearly in the paper.
From
G
and
γ
one computes
Rfrequency = log2 G / γ bits per site,
the information needed to locate the sites in the genome.
The information measured in the sites (Rsequence, bits per site)
starts at zero and
entirely by an evolutionary process
Rsequence
converges on
Rfrequency.
In the figure to the right,
Rfrequency is shown by the dashed line and
the evolving Rsequence is the green curve.
At 1000 generations the population was duplicated and in one
case selection continued (horizontal noisy green curve)
and in the other case selection was turned off
(exponentially decaying noisy red curve).
Thus the information gain depends on selection and it not blind and unguided.
The selection is based on biologically sensible criteria: having
functional DNA binding sites and not having extra ones. So Ev models
the natural situation.
You can try this experiment yourself
on any computer in less than 10 seconds (!)
by using the new
Rfrequency = 4.00 bits/site Rsequence ≈ 4 bits/site (fluctuating) So the information in the binding sites (Rsequence) evolves to matches the predicted value (Rfrequency). See the Guide to Evj for more details about running this program. |
Note that the size of the genome is determined by the number of functions required for survival in an environment. For example, the bacterium E. coli has about γ = 4,000 genes. It doesn't need more than that to survive in its environment. We know that parasites have fewer genes because they can use the nutrients from their host. So E. coli doesn't lose genes either and γ is fixed by the environment, as is the size of the genome. E. coli has a genome of about G = 4.6 million base pairs, each of which codes for roughly only one strand of mRNA. So Rfrequency = log2 4.6 x 106 / 4000 = 10.1 bits per site. This information is determined by the environment. The information in a verified set of E. coli genes is Rsequence = 10.35(+/-0.16) bits. So Rfrequency is within two standard deviations of Rsequence. The information from the environment becomes imbedded in the genome by natural selection. The Ev/Evj programs demonstrate this process. (For further details on this computation, see Anatomy of Escherichia coli Ribosome Binding Sites.)
132 141 148 158 164 174 181 191 201 208 214 223 232 240 248 256The differences between these numbers are:
9 7 10 6 10 7 10 10 7 6 9 9 8 8 8The coordinates reported in Montanez.Marks2010 are:
1 10 17 26 33 43 50 60 70 76 83 92 101 109 117 125The differences between these numbers are:
9 7 9 7 10 7 10 10 6 7 9 9 8 8 8As indicated in red, two of the sites were misplaced by one base. However, this error will not change the results as can be seen by noting that Ev is an entirely different program from Evj but they both give the same results and from repeat runs of either program with different initial random seeds.
As far as ev can be viewed as a model for biological processes in nature, it provides little evidence for the ability of a Darwinian search to generate new information.This is incorrect. They aren't looking at the right information. What they call "external knowledge" is Rfrequency and that represents information outside the genetic control system and in general outside or being directed from the the environment. But it is absolutely clear by looking at the sequene logo at the start of any Evj run that the information inside the genome starts at zero and then increases to the requesite amount by evolution.
Rather, it demonstrates that preexisting sources of information can be re-used and exploited, with varying degrees of efficiency, by a suitably designed search process, biased computation structure, and tuned parameter set.So that's exactly what the evolutionary process is doing - adapting the organism to the environment by capturing information about the environment in its genome.
Summary Aside from their propensity to veer away from the actual biological situation, the main flaw in this paper is the apparent misunderstanding of what Ev is doing, namely what information is being measured and that there are two different measures. The authors only worked with Rfrequency and ignored Rsequence. They apparently didn't compute information from the sequences. But it is the increase of Rsequence that is of primary importance to understand. Thanks to Chris Adami, we clearly understand that information gained in genomes reflects 'information' in the environment. I put environmental 'information' in quotes because it is not clear that information is meaningful when entirely outside the context of a living organism. An organism interprets its surroundings and that 'information' guides the evolution of its genome.
Response by Robert Marks The autopsy of a dissection of a vivisection: response to Schneider's response by Robert Marks (2011-09-03)
"We agree with Schneider that information is gained by the genome through extraction of it from the environment."Since this is what happens in nature the game is over. Saying that the mechanism is a perceptron, etc, just obscures the issues. Ev models what happens in nature as shown by my previous work on binding sites, Information Content of Binding Sites.
Other papers and ideas by Dembski have been evaluated previously on related pages:
![]()

Schneider Lab
origin: 2011 Feb 16
updated:
version = 1.07 of Montanez.Marks2010.html 2013 Nov 12
![]()