>



Next: Bibliography
Up: Evolution of Biological Information
Previous: DISCUSSION
I thank
Denise Rubens,
Ilya Lyakhov,
Herb Schneider,
Natasha Klar,
Bruce Shapiro,
Richard Dawkins,
Hugo Martinez
and
Karen Lewis
for comments on the manuscript,
and Frank Schmidt for pointing out that the
atrophy should be first order.
Figure 1:
Genetic sequence of a computer organism.
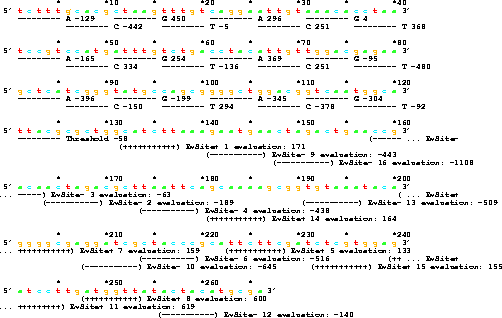
The organism has two parts, a weight matrix gene and a binding site region.
The gene for the weight matrix covers bases 1 through 125.
It consists of
6 segments 20 bases wide and one tolerance value 5 bases wide.
Each segment contains sequence specifying the weights for the four
nucleotides. For example, bases 1 to 5 contain tcttt.
Translating this to binary gives 1101111111, which is
the twos complement number for -129.
This is the weight for A in the first position of the matrix.
The 16 non-overlapping binding site locations were placed at random in the remaining
portion of the genome.
Evaluation by the weight matrix is indicated for each site.
For example site 1, covering positions 132 to 137,
catctt,
is evaluated as
-442 +296 -136 +251 +294 -92 = 171.
Since this is larger than the threshold (-58),
it is `recognized', and is marked with `+' signs.
Evaluations to determine mistakes are for the first 256 positions on the genome.
An extra 5 bases are added to the end, but not searched,
to allow the sequence logos in
Fig. 3 to have complete sequences available at all positions.
Mutations are applied to all positions in the genome, so the binding
sites and the weight matrix co-evolve.
The figure was generated with programs
ev,
evd
and
lister.
|
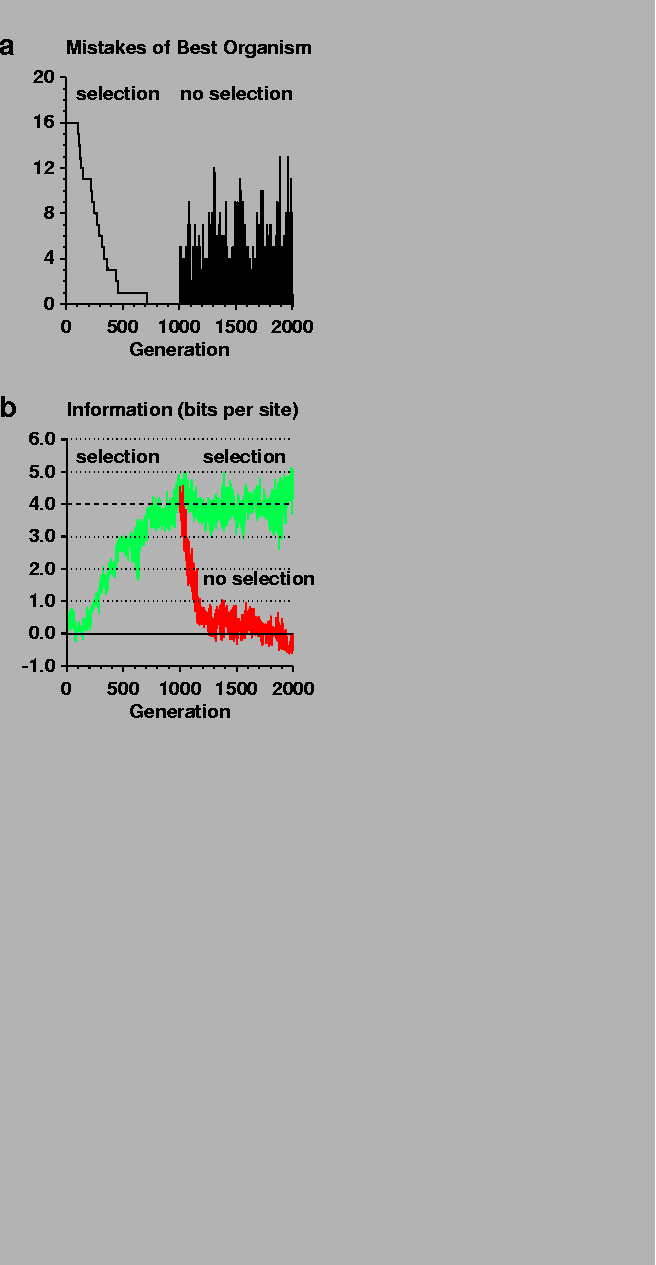
Figure 2:
Information gain by natural selection.
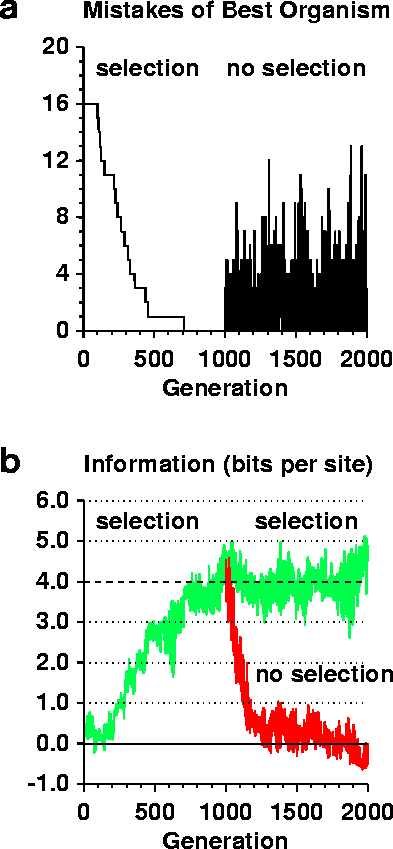
a, Number of mistakes made by the organism with the fewest mistakes
is plotted against the generation number.
At 1000 generations, selection was removed.
Because of the initial random number arbitrarily
chosen for this particular simulation (0.3),
the initial best organism only made mistakes in missing the 16 sites,
but this is generally not the case.
(Displaying the best organism, which is most likely to survive,
is a form of selection that does not affect the simulation.)
b,
The information content at binding sites
(
Rsequence)
of the organism making the fewest mistakes
is plotted against generation number.
Selection for organisms making the fewest mistakes
was applied from generation 0 to 2000 (top curve, green).
The simulation was then reset to the state at 1000 generations
and rerun without selection (bottom curve, red).
The dashed line shows the information predicted,
Rfrequency = 4 bits,
given the size of the genome and the number of binding sites.
|
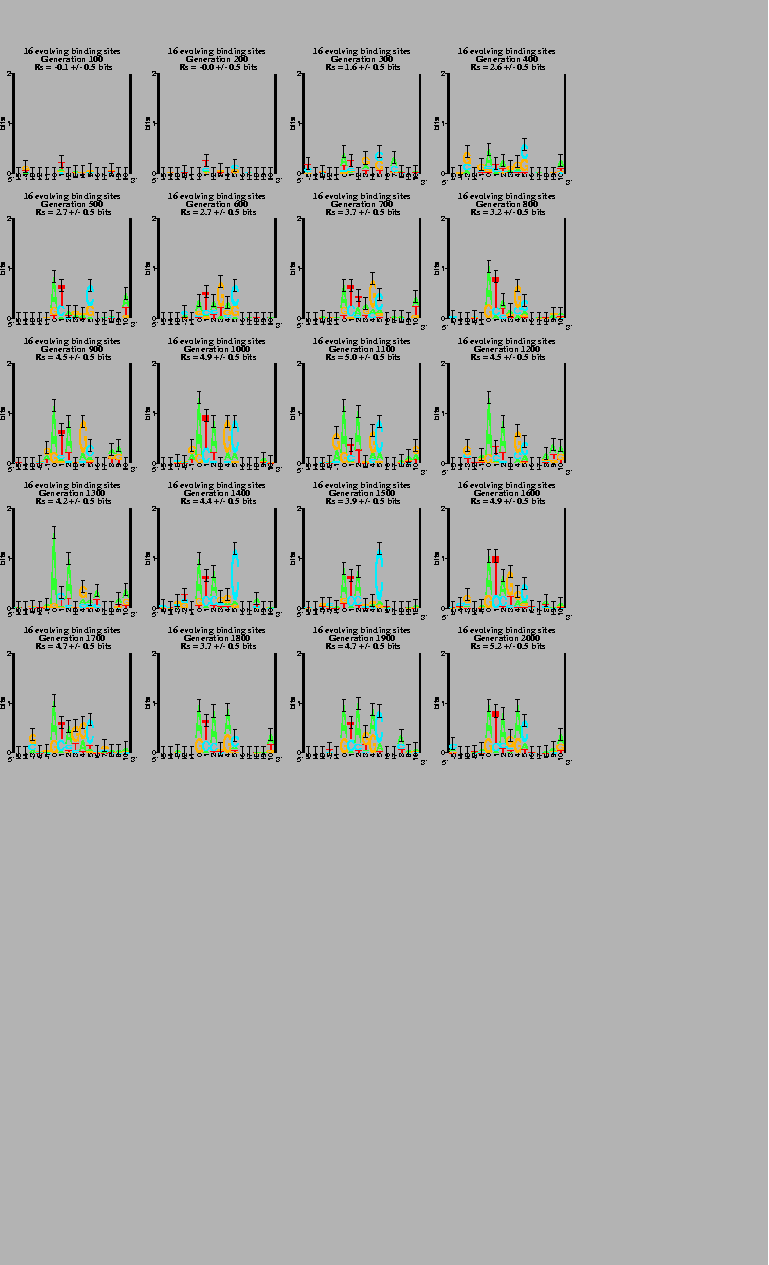
Figure 3:
Sequence logos showing evolution of binding sites.
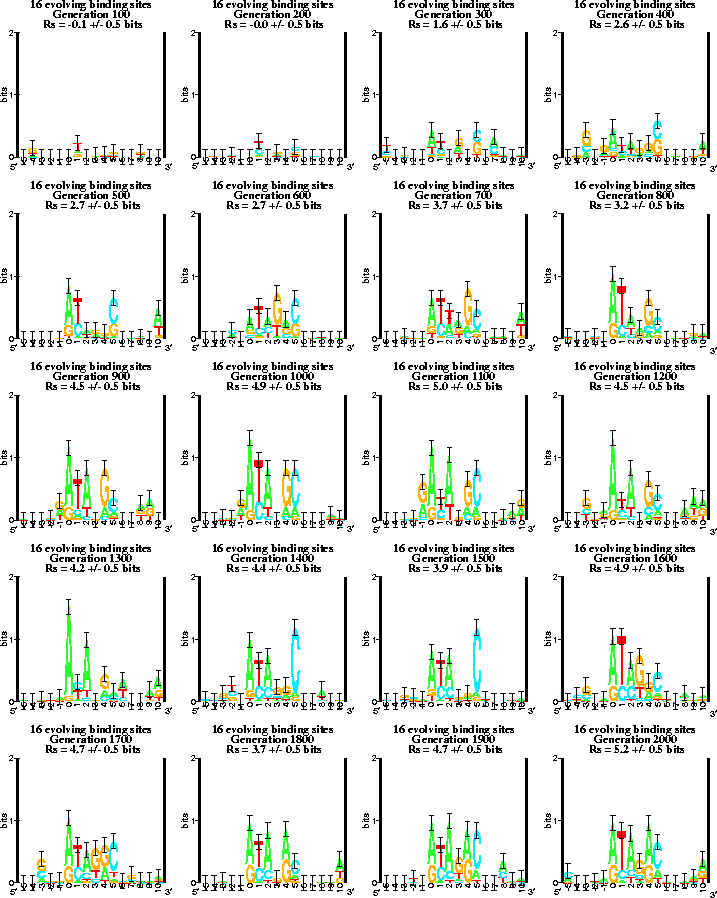
A
sequence logo
shows the information content at a set of binding
sites by a set of stacks of letters [5].
The height of each stack
is given in bits, and the sum of the heights is the total information
content,
Rsequence.
Within each stack the relative heights of each
letter are proportional to the frequency of that base at that position,
f(b,l).
Error bars indicate likely variation caused by the small sample
size [4], as seen outside the sites,
which
cover positions 0 to 5.
The complete movie is available at
http://www.lecb.ncifcrf.gov//paper/ev/movie.
|
Next: Bibliography
Up: Evolution of Biological Information
Previous: DISCUSSION
Tom Schneider
2001-11-07