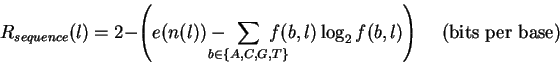Thomas D. Schneider1
version = 1.50 of oxyr.tex 1999 October 12
Methods in Enzymology:
RNA Polymerase and Associated Factors,
Part B, Volume 274, pages 445-455, 1996
Edited by Sankar Adhya
Supplemental information:
http://www.lecb.ncifcrf.gov//how.to.read.sequence.logos/index.html
running title:
Reading Sequence Logos
Information theory was introduced in the late 1940s by Claude Shannon for the study of communications systems[1,2]. With this mathematical tool, information passing through a telephone or computer data line can be measured and compared to the theoretical limit of the line, called the channel capacity. The information measure is given in units of bits per second, where one bit is the choice between two equally likely possibilities. Surprisingly, one of Shannon's theorems states that as long as the channel capacity is not exceeded, the communications may have as few errors as desired[3]. An example of the practical use of this result is the clear music of compact discs (CDs) which are specially coded to protect against noise. Cleaning instructions for CDs say that they should be wiped in a radial direction. If any scratches are introduced, they will have less effect on the coding, which runs in concentric circles and is capable of correcting up to 4000 data bit errors[4].
Evolutionary conservation indicates the functional importance of biological structures, so a robust and precise measure for conservation is necessary for empirical studies. The measurement of conservation in bits is uniquely suited to this task because, unlike any other measure, bits are additive and consistent from one system to the next. Bits provide a universal scale for measuring biological conservation not only in DNA and RNA but also for proteins and other macromolecules[5,6].
OxyR is a tetrameric protein that binds to the DNA of several promoters in Escherichia coli and activates transcription of genes encoding antioxidant enzymes[7,8]. The initial investigation of the DNA-binding sites by Tartaglia et al. showed that OxyR binds to a large region of DNA, but the consensus sequences obtained were weak and sparse, making the sites difficult to characterize.
In conjunction with the information analysis described here, recent experimental work on this protein[9] clarified the way in which OxyR binds to DNA sequences. This would not have been possible using a consensus sequence.
Materials and Methods
Sequences
When possible, wild-type OxyR DNA binding site sequences[7,9] were obtained from GenBank 83. The following list includes the site name, accession number, and coordinate of the central (zero) base: oxyR, J04553, 163; katG, M21516, 68; ahpC, D13187, 116; dps, X69337, 202; gorA, U00039, 60491; Mu mom, V01463, 59; S. typhimurium orf, (not in GenBank) 29. Both wild-type and the random sequences that bound to OxyR are given elsewhere[9].
Programs and Data
Information analysis
and the sequence logo technique
were performed as described
previously [5,10,11].
A primer on information theory is available
on the internet at
https://alum.mit.edu/www/toms/ftp/primer.ps.
Programs (written in Pascal[12])
and data are available by anonymous ftp from
https://alum.mit.edu/www/toms/ftp/
or via the World Wide Web site
http://www-lmmb.ncifcrf.gov/![]() toms/.
toms/.
The programs for constructing sequence logos and performing other tasks were used in approximately this order:
The aligned listing and sequence logos were printed on a Tektronix Phaser 140 inkjet printer.
Sequence Logos
The sequence logos[10,11] in Fig. 2 summarize the data in a set of aligned sequences such as Fig. 1. The height of a stack of letters is the sequence conservation measured in bits of information according to equation (1). The height of each letter within a stack is proportional to its frequency at that position in the binding site. The letters are sorted, with the most frequent on top. The cosine wave represents the twist of B-form DNA. Wave peaks are all on one face of the DNA and represent the major groove facing the protein. Error bars indicate the variability of a comparable number of random sequences[5].
Results and Discussion
Analysis of OxyR Sequence Logos for DNA Binding Face
Wild-type OxyR binding site sequences
were aligned
(Fig. 1)
and information analysis was used to generate the
sequence logo in Fig. 2a.
The logo
shows a correlation between the strongest sequence conservation,
as given by the heights of the stacks of letters,
and the face of B-form DNA, as given by the cosine wave.
The same correlation is seen in
other proteins
(![]() cI/cro,
cI/cro,
![]() O,
434 cI/cro,
ArgR,
CRP,
TrpR,
FNR
and
LexA,
see figure 6 of Papp et al.[11]).
The extent of DNase I footprinting[7]
and this correlation
suggests that OxyR binds to one face of B-form DNA
in 4 successive major grooves.
O,
434 cI/cro,
ArgR,
CRP,
TrpR,
FNR
and
LexA,
see figure 6 of Papp et al.[11]).
The extent of DNase I footprinting[7]
and this correlation
suggests that OxyR binds to one face of B-form DNA
in 4 successive major grooves.
A second line of evidence that can be read from the sequence logo
also supports this model.
When a protein is in contact with a major groove,
the two base pairs and their two orientations
can be distinguished,
as recognized by Seeman
et al[17,11],
so the protein is capable of ``choosing''
one of the four possibilities:
A=T,
T=A,
C![]() G,
or
G
G,
or
G![]() C.
This can be explained with the help of
Fig. 3,
which depicts the two base pairs.
The possible chemical contacts for T=A
in the major groove are (reading from left to right):
methyl group,
hydrogen acceptor,
hydrogen donor
and
hydrogen acceptor,
or
T:Me-a-d-a:A for short.
This can easily be distinguished from the
complementary pattern of
A=T, which is A:a-d-a-Me:T. Likewise
C:(blank)-d-a-a:G
is distinguishable from
G:a-a-d-(blank):C.
Finally, GC/CG can be distinguished from AT/TA.
C.
This can be explained with the help of
Fig. 3,
which depicts the two base pairs.
The possible chemical contacts for T=A
in the major groove are (reading from left to right):
methyl group,
hydrogen acceptor,
hydrogen donor
and
hydrogen acceptor,
or
T:Me-a-d-a:A for short.
This can easily be distinguished from the
complementary pattern of
A=T, which is A:a-d-a-Me:T. Likewise
C:(blank)-d-a-a:G
is distinguishable from
G:a-a-d-(blank):C.
Finally, GC/CG can be distinguished from AT/TA.
This choice of 1 possibility in 4 can be made
with 2 bits of information.
(This is calculated as
![]() bits.
For further explanation, see Pierce[2] or
the primer on information theory whose location is given in
Materials and Methods.)
Completely conserved
positions in the major groove are described by 2 bits
and this is the highest point on the vertical scale of the sequence logos.
It is easiest to think of a bit as a knife slice that dissects
the bases in
Fig. 3.
A horizontal slice is the first bit and a vertical slice is the second one.
The first bit determines whether the base is
above or below the slice
and the second bit determines whether
it is to the left or the right.
Because they are at right angles to one another,
the slices provide independent choices
and no more than 2 bits are needed to specify a single base.
For example, ``top, left'' selects the T.
The average number of bits needed to describe the observed
frequency of bases is the information content
or sequence conservation.
Because it is an average it does not need to be an integer
and
so the heights of the letter stacks in the sequence logo are real
numbers.
Sequence conservation in the major groove can range anywhere between
0 and 2 bits depending on the strength of the contacts involved,
as seen in figure 6 of Papp et al.[11].
Just because it is possible for sequence conservation
to be as high as 2 bits from the major groove does not mean that the
protein will evolve to that high a value.
The important factors are the total sequence conservation[5]
and the
coding of the binding site that distinguishes it from other sequences.
bits.
For further explanation, see Pierce[2] or
the primer on information theory whose location is given in
Materials and Methods.)
Completely conserved
positions in the major groove are described by 2 bits
and this is the highest point on the vertical scale of the sequence logos.
It is easiest to think of a bit as a knife slice that dissects
the bases in
Fig. 3.
A horizontal slice is the first bit and a vertical slice is the second one.
The first bit determines whether the base is
above or below the slice
and the second bit determines whether
it is to the left or the right.
Because they are at right angles to one another,
the slices provide independent choices
and no more than 2 bits are needed to specify a single base.
For example, ``top, left'' selects the T.
The average number of bits needed to describe the observed
frequency of bases is the information content
or sequence conservation.
Because it is an average it does not need to be an integer
and
so the heights of the letter stacks in the sequence logo are real
numbers.
Sequence conservation in the major groove can range anywhere between
0 and 2 bits depending on the strength of the contacts involved,
as seen in figure 6 of Papp et al.[11].
Just because it is possible for sequence conservation
to be as high as 2 bits from the major groove does not mean that the
protein will evolve to that high a value.
The important factors are the total sequence conservation[5]
and the
coding of the binding site that distinguishes it from other sequences.
In contrast to the major groove,
contacts in the minor groove of B-form DNA allow both orientations of each kind
of base pair so that
rotations about the dyad axis cannot easily be distinguished.
This is because
from the minor groove
C![]() G
appears nearly identical to
G
G
appears nearly identical to
G![]() C
and
A=T
appears identical to
T=A.
Fig. 3 shows that
C
C
and
A=T
appears identical to
T=A.
Fig. 3 shows that
C![]() G in the minor groove
has the chemical moiety pattern
a-d-a,
which is, to a good first approximation,
identical to the pattern of the complementary orientation.
The hydrogen donor N2 of G
is almost exactly on the dyad axis (the dashed line) and
so its position does not change much in the complement. Hydrogen atoms held in
a hydrogen bond vibrate vigorously and probably make such a fine
positional
distinction difficult because they vibrate almost independently
of the donor and acceptor[18].
The base pair
A=T
has
a-(blank)-a,
which is
identical in the other orientation.
So A=T can be distinguished from C
G in the minor groove
has the chemical moiety pattern
a-d-a,
which is, to a good first approximation,
identical to the pattern of the complementary orientation.
The hydrogen donor N2 of G
is almost exactly on the dyad axis (the dashed line) and
so its position does not change much in the complement. Hydrogen atoms held in
a hydrogen bond vibrate vigorously and probably make such a fine
positional
distinction difficult because they vibrate almost independently
of the donor and acceptor[18].
The base pair
A=T
has
a-(blank)-a,
which is
identical in the other orientation.
So A=T can be distinguished from C![]() G in the minor groove
only by a donor contact to the N2 or by a physical probe which
blocks the N2 (in the blank at the black dot).
Because both of these are close to the dyad axis
only the horizontal knife slice works for the minor groove.
G in the minor groove
only by a donor contact to the N2 or by a physical probe which
blocks the N2 (in the blank at the black dot).
Because both of these are close to the dyad axis
only the horizontal knife slice works for the minor groove.
Because
only 2 of the 4 possibilities can be distinguished,
when a B-form minor groove is probed by a protein no more
than 1 bit of information (
![]() bit) can be obtained.
That is, positions with more than 1 bit of information
are likely to represent major grooves facing the protein
or, if they do represent minor groove contacts,
then the DNA is probably not B-form[11].
In the OxyR sequence logo, positions
bit) can be obtained.
That is, positions with more than 1 bit of information
are likely to represent major grooves facing the protein
or, if they do represent minor groove contacts,
then the DNA is probably not B-form[11].
In the OxyR sequence logo, positions ![]() ,
,
![]() and
and ![]() are conserved by more than 1 bit of information, so
these positions probably represent major grooves facing the protein.
This is consistent with the correlation between sequence conservation
and the face of the DNA discussed earlier.
are conserved by more than 1 bit of information, so
these positions probably represent major grooves facing the protein.
This is consistent with the correlation between sequence conservation
and the face of the DNA discussed earlier.
After this prediction was made, it was confirmed by hydroxyl radical footprinting and missing base experiments[9].
Prediction of Specific OxyR DNA Contacts
by ``Reading'' the Sequence Logo
Because position 0 in the wild-type sequence logo
shows equally likely A and T (Fig. 2a),
that position may represent a contact from OxyR which
collides with the minor groove N2 of G (ref.[17]),
allowing only
A=T
and
T=A,
and disallowing
C![]() G entirely.
This prediction was supported by methylation interference data
which indicate that methylation at N3 of A blocks binding
(Fig. 2a).
G entirely.
This prediction was supported by methylation interference data
which indicate that methylation at N3 of A blocks binding
(Fig. 2a).
The logo also shows that
positions +4, +7 and +13 are predominantly
either A or G
(T or C at the negative coordinates), which suggests a contact by OxyR to the
major groove N7 group because only the N7 acceptor is
conserved in an A
![]() G transition
(T:Me-a-d-
G transition
(T:Me-a-d-![]() :A
matches C:(blank)-d-a-
:A
matches C:(blank)-d-a-![]() :G
only in the last moiety, see Fig. 3).
This is also confirmed by methylation interference data, but
the disproportionate frequencies of these bases suggests that other
contacts or effects are also involved.
:G
only in the last moiety, see Fig. 3).
This is also confirmed by methylation interference data, but
the disproportionate frequencies of these bases suggests that other
contacts or effects are also involved.
Position -15 is mostly T or G (A or C at +15),
suggesting a weak contact to the
major groove A-N6 or C-N4 and/or T-O4 and G-O6 groups.
These contacts could be conserved in
T
![]() G transversions because they shift by only
G transversions because they shift by only
![]() Å.
(T:Me-
Å.
(T:Me-![]() -
-![]() -a:A
matches G:a-
-a:A
matches G:a-![]() -
-![]() -(blank):C
in the middle two moieties,
see Fig. 3).
This kind of contact
is likely to be at position -6 (and +7) of CRP sites[11],
where the crystal structure shows that
Arg180 donates hydrogen bonds to O6 and N7 of
guanine[19].
The preferred binding order G > T > A
-(blank):C
in the middle two moieties,
see Fig. 3).
This kind of contact
is likely to be at position -6 (and +7) of CRP sites[11],
where the crystal structure shows that
Arg180 donates hydrogen bonds to O6 and N7 of
guanine[19].
The preferred binding order G > T > A ![]() C is directly
visible in the CRP sequence logo[11],
and is confirmed by mutations[20,21,19].
Apparently when G is replaced by T in a CRP site, the T-O4 contact
is used instead of G-O6, but the G-N7 contact is lost.
This accounts for the binding order
except for the A.
Substitution by A would break the G-O6 contact
but would maintain the N7 contact.
The binding order
T > A
suggests that
the O4 contact is stronger than the N7 contact[20],
even though when T
C is directly
visible in the CRP sequence logo[11],
and is confirmed by mutations[20,21,19].
Apparently when G is replaced by T in a CRP site, the T-O4 contact
is used instead of G-O6, but the G-N7 contact is lost.
This accounts for the binding order
except for the A.
Substitution by A would break the G-O6 contact
but would maintain the N7 contact.
The binding order
T > A
suggests that
the O4 contact is stronger than the N7 contact[20],
even though when T
![]() G,
the O4/O6 contact shifts by
G,
the O4/O6 contact shifts by
![]() Å whereas
the N7 contact does not move at all.
Å whereas
the N7 contact does not move at all.
Other positions in the OxyR binding sites show methylation interference that do not have apparent correlation to the observed sequence conservation. This could be because the sequence conservation was derived from only a few sequences and so is noisy, as indicated by the large error bars. Alternatively, OxyR may pass close to the DNA at some points but not make actual contact unless the DNA is abnormally methylated. Other effects, such as DNA bending or twisting, also might account for these discrepancies.
A more subtle piece of evidence can be found in the overall shape
of the sequence logo.
Notice how the stack heights at positions
-4, -5, and -7follow along under the cosine wave
(Fig. 2a).
This effect can be observed in other sequence logos,
in particular
![]() cI/cro,
cI/cro,
![]() O,
434 cI/cro,
CRP,
FNR
and
LexA[11].
It can be explained by the geometry of a globular protein approaching
the cylindrical DNA.
During the process of finding the binding sites the protein
moves toward and away from the DNA[22].
Contacts at the center of the DNA cylinder are closest to the protein
and so should be the easiest to evolve.
Contacts become progressively more difficult to make
as the
approach is made further off axis
(Fig. 4).
If one were to rotate a DNA molecule
on its long axis, a point on its surface
would cycle between being visible and not visible.
This naturally results in a cosine function of accessibility
along a linear DNA molecule.
If we define accessibility as
a cosine function that runs from 0 to 1 bit in the minor groove
and
another cosine function that runs
from 0 to 2 bits in the major groove,
then the sum of these two functions is the cosine wave drawn on the logo.
The correlation between sequence conservation
and accessibility is
consistent with the proposal that
positions
O,
434 cI/cro,
CRP,
FNR
and
LexA[11].
It can be explained by the geometry of a globular protein approaching
the cylindrical DNA.
During the process of finding the binding sites the protein
moves toward and away from the DNA[22].
Contacts at the center of the DNA cylinder are closest to the protein
and so should be the easiest to evolve.
Contacts become progressively more difficult to make
as the
approach is made further off axis
(Fig. 4).
If one were to rotate a DNA molecule
on its long axis, a point on its surface
would cycle between being visible and not visible.
This naturally results in a cosine function of accessibility
along a linear DNA molecule.
If we define accessibility as
a cosine function that runs from 0 to 1 bit in the minor groove
and
another cosine function that runs
from 0 to 2 bits in the major groove,
then the sum of these two functions is the cosine wave drawn on the logo.
The correlation between sequence conservation
and accessibility is
consistent with the proposal that
positions
![]() ,
,
![]() ,
and
,
and
![]() are read from the major groove.
are read from the major groove.
To summarize, there are at least four interrelated techniques that can be used to read a sequence logo:
Analysis of Synthetic OxyR Binding Sites
A ``randomization''
experiment[23,11,24]
was performed
in which OxyR protein was used to gel shift 30 base pair
equi-probable random sequences[9].
Unfortunately this gave a dismal logo (Fig. 2b), possibly because
the protein was prevented from binding properly by the
flanking constant
sequence of the vector.
The high conservation at the ends (![]() and
and ![]() )
comes more from one side of the cloning
sites and so may represent an
artifact.
2
Still, some correlation with Fig. 2a is visible in the logo,
in particular the A preference at
)
comes more from one side of the cloning
sites and so may represent an
artifact.
2
Still, some correlation with Fig. 2a is visible in the logo,
in particular the A preference at ![]() ,
the T preference at
,
the T preference at ![]() ,
and
the G preference at
,
and
the G preference at ![]() .
But other positions are just as conserved and do not reflect the wild-type
sequences.
.
But other positions are just as conserved and do not reflect the wild-type
sequences.
To clarify this situation,
the randomization experiment was repeated with 45 base pair
equi-probable random sequences which were then aligned by an information theory
technique using the malign.p program (Fig. 2c)[9].
Only some of the patterns evident in Fig. 2a
were confirmed by this experiment,
whereas others became more predominant.
The T or C at position ![]() closely reflects
the wild-type sequences there (7 Cs, 5 Ts, 1 G, 1 A).
The conservation at
closely reflects
the wild-type sequences there (7 Cs, 5 Ts, 1 G, 1 A).
The conservation at ![]() and
and ![]() increased but positions 0 and
increased but positions 0 and ![]() barely increased.
The wild-type cluster at
barely increased.
The wild-type cluster at ![]() ,
,
![]() and perhaps
and perhaps ![]() did materialize
but not strongly.
The almost insignificantly weak preferences for
A at
did materialize
but not strongly.
The almost insignificantly weak preferences for
A at ![]() ,
T at
,
T at ![]() ,
and T at
,
and T at ![]() of wild-type appear amplified.
Additional conservation not seen previously appeared at
of wild-type appear amplified.
Additional conservation not seen previously appeared at
![]() (?),
(?),
![]() ,
,
![]() ,
,
![]() and
and
![]() .
The reason for these quantitative
discrepancies between the wild-type
sequence logo and the logo from experimentally selected sites is unknown,
but might be accounted for by the small sample sizes.
.
The reason for these quantitative
discrepancies between the wild-type
sequence logo and the logo from experimentally selected sites is unknown,
but might be accounted for by the small sample sizes.
Another difficulty with this kind of experiment is that it always contains at least one unknown parameter, the stringency of selection. If the concentration of OxyR protein were large, then its non-specific binding should cause more DNA sequences to shift in the gel. This would lead to a sequence logo with a low information content relative to the natural sequences. However, a low concentration of OxyR protein should lead to a much higher measured information content, perhaps higher than is naturally found. This is also a danger in amplification protocols such as SELEX[25].
To counter this,
the protein concentration could be adjusted
so that the total information content from the randomization experiment
matches that of the natural sequences. Presumably the two logos
would then look the same.
Were they the same for the experiment that was done?
The area under the
logo (
Rsequence) was
![]() bits for the 7 wild-type
sequences and their complements
(Fig. 2A)
but was
bits for the 7 wild-type
sequences and their complements
(Fig. 2A)
but was
![]() bits
for the 16 sequences and their complements in the 45mer experiment
(Fig. 2C).
These can be compared by a two-tailed Student's t test[16].
Because both the sequences and their complements were used,
the two halves of the sequence logo are not independent and the test
must be done with half-sites.
This reduces each mean by a factor of 2 and reduces
the variance by a factor of 2 so the standard deviations reduce by
bits
for the 16 sequences and their complements in the 45mer experiment
(Fig. 2C).
These can be compared by a two-tailed Student's t test[16].
Because both the sequences and their complements were used,
the two halves of the sequence logo are not independent and the test
must be done with half-sites.
This reduces each mean by a factor of 2 and reduces
the variance by a factor of 2 so the standard deviations reduce by
![]() .
The number of samples in the half-site set
remains at 14 and 32 respectively.
The t test on the half-sites shows that
the natural sites are significantly different
in information content from the experimental set
(t=2.7 with 44 degrees of freedom, p < 0.02).
Unfortunately this criterion for matching the wild-type binding
sites was not met.
.
The number of samples in the half-site set
remains at 14 and 32 respectively.
The t test on the half-sites shows that
the natural sites are significantly different
in information content from the experimental set
(t=2.7 with 44 degrees of freedom, p < 0.02).
Unfortunately this criterion for matching the wild-type binding
sites was not met.
Even so, it is clear that the sequence conservation is not uniformly proportional across the two logos. One possibility that might account for the observed conservation at the edges of the experimentally derived sites is that OxyR is still affected by the constant flanking sequences of the cloning vector and binds to one or the other side in some cases. The discrepancy may also reflect different conditions between in vivo evolutionary factors and the in vitro gel shift experiment. For example, no spermidine was used in the gel shift experiment[9], yet it is well known that spermidine is important for precise recognition by other DNA binding proteins[26,27]. Comparison of the wild-type sequence logo to a series of random gel shift sequence logos might be used to determine precisely what the in vivo binding conditions are.
Usefulness of Sequence Logos Versus Consensus Sequences
The case of OxyR demonstrates the usefulness of sequence logos as a replacement for consensus sequences. The pattern bound by the protein is difficult to detect by eye (Fig. 1), and no agreement could be found for a consensus sequence. In contrast, the sequence logos are created automatically and without any ambiguity. They show clear and easily interpretable patterns. Because information theory is quantitative, statistical tests can be applied to collections of binding site sequences.
A consensus replaces the natural frequencies of bases with arbitrarily chosen ones [28]. For example, in our data set[11], CRP position -6has 2 As, 2 Cs, 44 Gs and 10 Ts. Taking the consensus alters this to 100% G and 0% of the other bases. When the consensus sequence G was chosen by this method, a subtle pattern of sequence conservation was lost[29,30,31,32,,33,21,19]. That the T contact occurs naturally apparently went unrecognized until the present work, although the mechanism of the contact was already understood from mutations[20].
The art of predicting specific base contacts is well known[17] but the pervasive use of consensus sequences in the modern molecular biology literature has prevented full use of the available sequence data. In the case of CRP discussed earlier, the subtle G to T switch, which probably destroys one hydrogen bond while keeping the other, was missed because only the G was retained in the consensus model. In contrast, because it visually displays the relevant information in a compact, quantitative form, the sequence logo allows direct interpretation of the data and leads to specific predictions that can guide experimentation. Further, when anomalies appear, the logo displays them so blatantly that new phenomena are revealed[11]. Even correlations between positions in a binding site[15] could be presented in a three-dimensional sequence logo, but software to generate this display has not been written yet.
As seen by the sequence logo, OxyR does not have a special kind of binding site as has been suggested[7]. OxyR merely happens to have a long binding site with low overall information content, so it tends to have low sequence conservation per position. As a consequence of the Second Law of Thermodynamics, DNA-protein contacts tend to spread out over the available surface on an evolutionary time scale. With at least 4 major grooves and three minor grooves to make contacts in, OxyR can ``afford'' to have many small contacts. Paradoxically, a mathematically rigorous theorem shows that having many small contacts like those used by OxyR can improve sequence discrimination[34]. Furthermore, many binding sites are like OxyR in that they have variations in their information content at different positions. This is immediately apparent upon inspection of splice junction sequence logos[15] and the ``gallery'' of DNA recognition sequence logos[11].
The arbitrary and artificial distinctions between strong and weak binding sites, between the ``core'' and the periphery of a site and between the inside and outside of binding site ``boxes'' that have been fostered by the use of consensus sequences are eliminated when one adopts the concept that sequence conservation is a real number that can be measured precisely in bits of information.
Summary
DNA sequences to which the OxyR protein binds under oxidizing conditions were analyzed by the sequence logo method, a quantitative graphic technique based on information theory. A sequence logo shows both the sequence conservation and the frequencies of bases at each position in a site. Unlike the consensus sequence, the sequence logo analysis revealed that OxyR should bind to four major grooves of DNA. This was later confirmed by experiments. Detailed interpretation of the sequence logo also allowed the prediction of likely major and minor groove OxyR-DNA base contacts, consistent with available experimental results. Because the sequence logo shows the original base frequencies in a clear, easily interpreted graphic that does not distort the data, highly refined analysis of binding site contacts becomes easy. Not only can these methods be applied to any DNA sequence binding site, they can also be applied to sites on RNA and proteins.
Acknowledgments
I thank Paul N. Hengen, Denise Rubens, Paul A. Smith, R. Michael Stephens, and R. E. Wolf for useful comments on the manuscript.
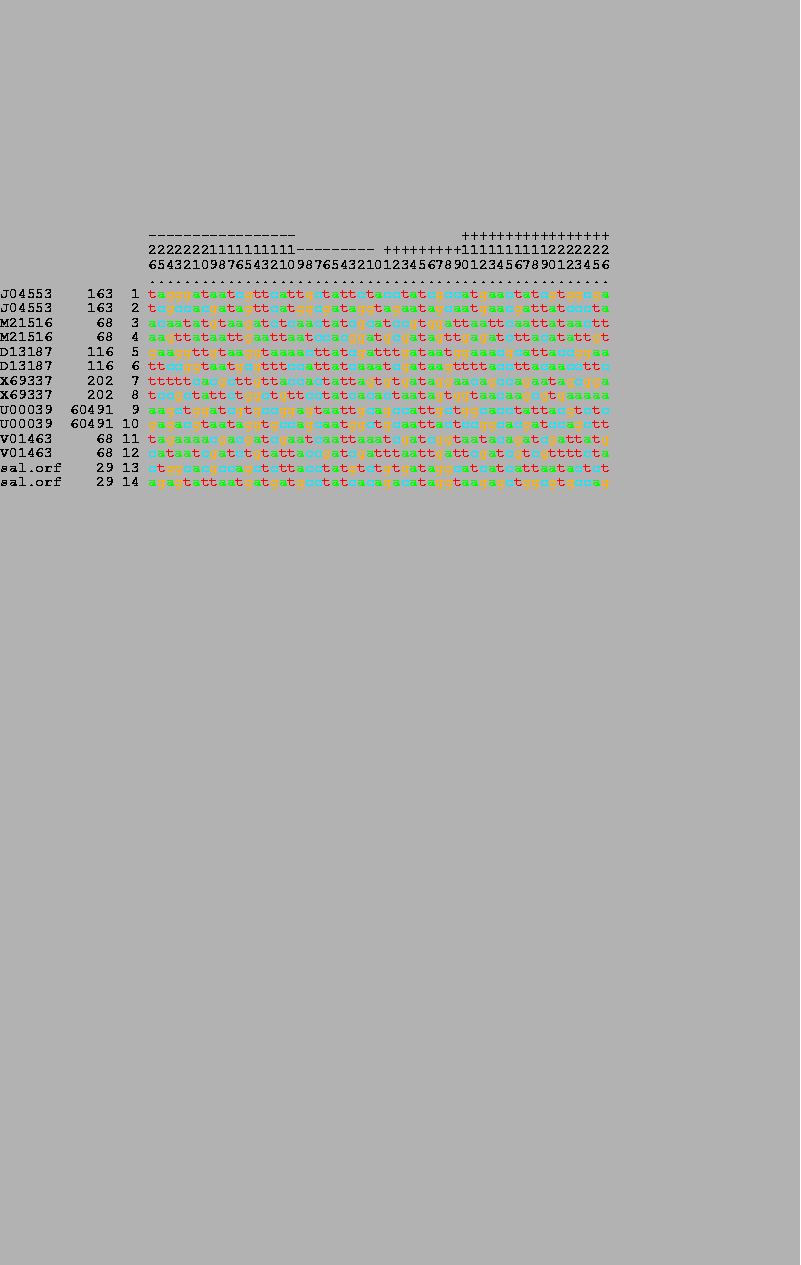
7 OxyR wild-type binding sites (odd numbers) and their complementary sequences (even numbers) were listed by the alist program. The numbers in the bar on the top are read vertically and give the position in the binding site, running from -26 to +26. The GenBank accession number, the coordinate of the zero base, and the number of each sequence are given on the left-hand side of the figure. |
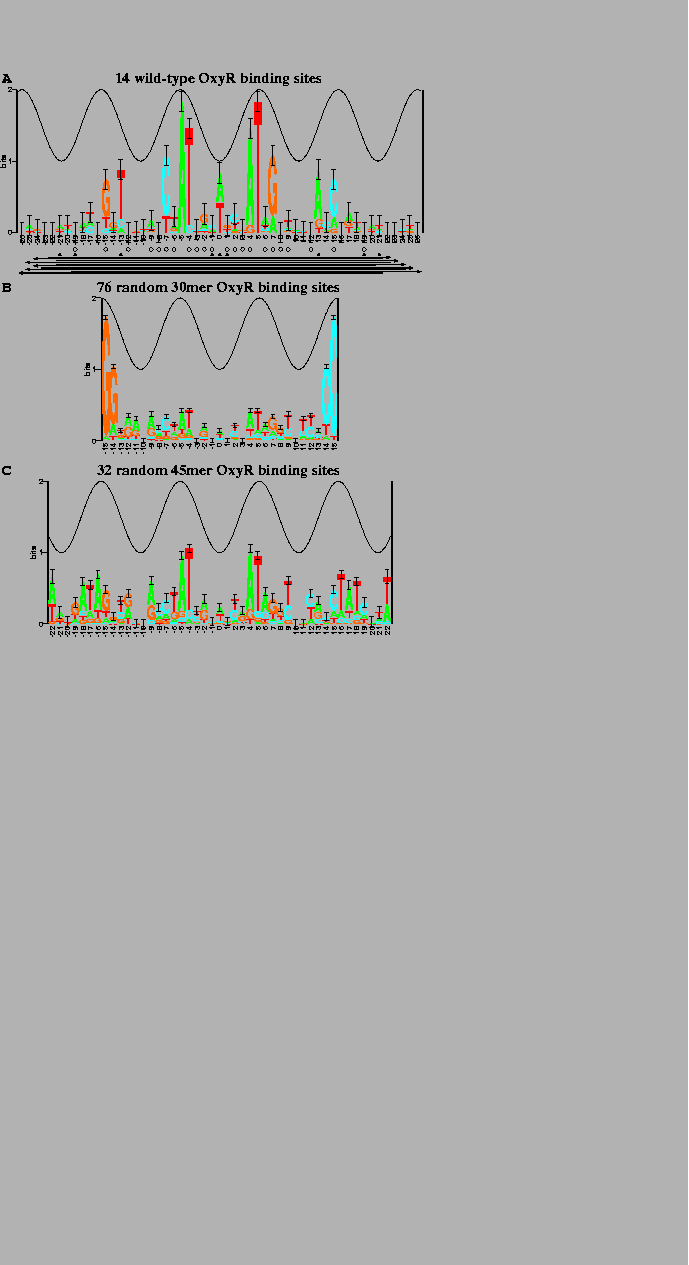
(A) 7 OxyR wild-type binding sites and their complementary sequences. The total sequence conservation, obtained by adding together the stack heights to determine the ``area'' under the logo (equation (2)), is |
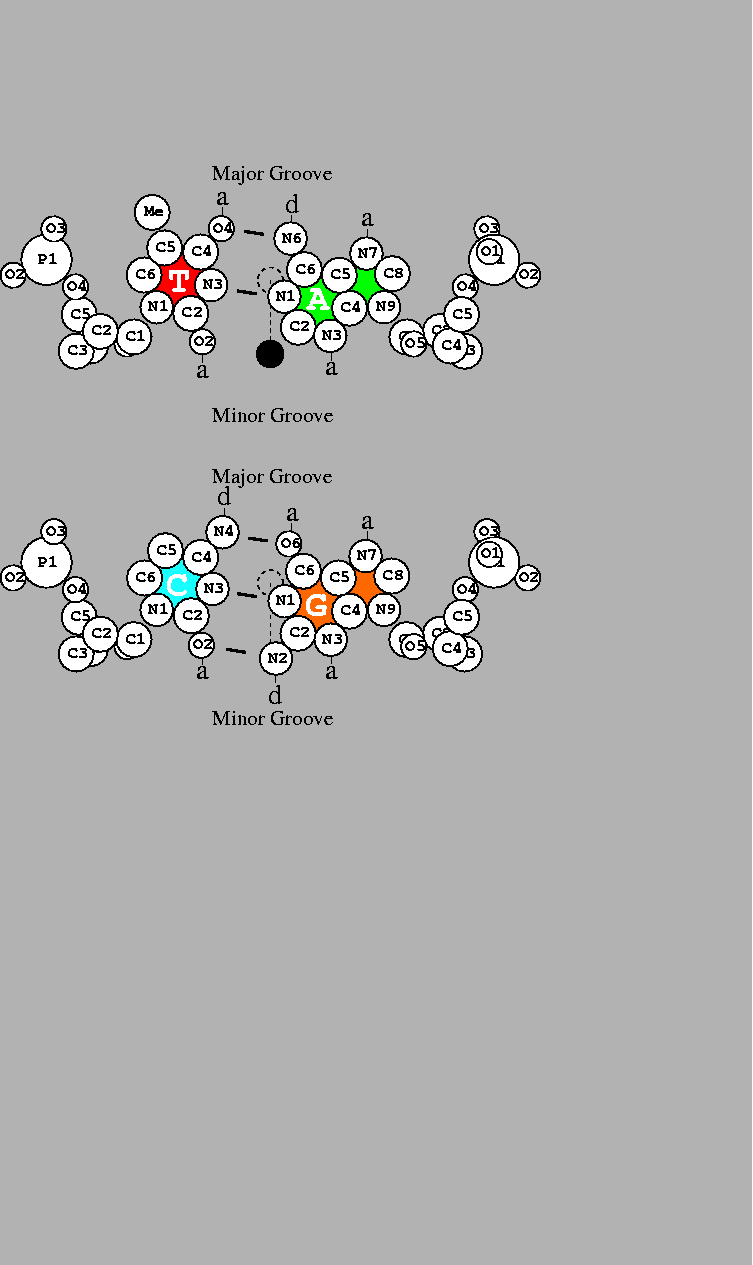
DNA base pairs drawn by the dnag program using coordinates for B-DNA [36] with atomic radii [37]. Short line segments indicate hydrogen bonds between the bases. a: acceptor of hydrogen bond; d: donor of hydrogen bond. The scale is shown by a 1Å diameter dashed circle placed on the helical axis. The two-fold dyad axis is indicated by a dashed line. Rotation by 180 |