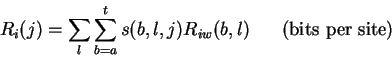Peter K. Rogan,1
2
![]() Brian M. Faux,*
Brian M. Faux,*![]()
and
Thomas D. Schneider
3
version = 1.73 of rfs.tex 1999 August 12
Abbreviated Title:
Information at human splice site mutations
Human Mutation 12: 153-171 (1998)
ABSTRACT
Splice site nucleotide substitutions
can be analyzed by comparing the individual information
contents (Ri, bits)
of the normal and variant splice junction sequences
[Rogan and Schneider, 1995].
In the present study, we related
splicing abnormalities
to
changes in Ri values of 111 previously
reported splice site substitutions in 41 different genes.
Mutant donor and acceptor sites have significantly less
information than their normal counterparts.
With one possible exception, primary
mutant sites with < 2.4 bits were not spliced.
Sites with Ri values ![]() bits
but less than the corresponding natural site usually
decreased but did not abolish
splicing.
Substitutions that
produced small changes in Riprobably do not impair splicing and are often polymorphisms.
The Ri values of activated
cryptic sites were generally comparable to
or greater than those of the corresponding natural splice sites.
Information analysis revealed
pre-existing
cryptic splice junctions that are used instead of the mutated
natural site.
Other cryptic sites were created or strengthened by sequence
changes that simultaneously altered the natural site.
Comparison between normal and mutant
splice site Ri values distinguishes substitutions that
impair splicing from those which do not,
distinguishes null alleles from those
that are partially functional,
and detects activated cryptic splice sites.
bits
but less than the corresponding natural site usually
decreased but did not abolish
splicing.
Substitutions that
produced small changes in Riprobably do not impair splicing and are often polymorphisms.
The Ri values of activated
cryptic sites were generally comparable to
or greater than those of the corresponding natural splice sites.
Information analysis revealed
pre-existing
cryptic splice junctions that are used instead of the mutated
natural site.
Other cryptic sites were created or strengthened by sequence
changes that simultaneously altered the natural site.
Comparison between normal and mutant
splice site Ri values distinguishes substitutions that
impair splicing from those which do not,
distinguishes null alleles from those
that are partially functional,
and detects activated cryptic splice sites.
keywords: information theory, mRNA splicing, donor, acceptor, cryptic, mutation, polymorphism, walker
INTRODUCTION
Mutations at splice sites make a significant contribution to human genetic disease, since approximately 15% of disease-causing point mutations affect pre-mRNA splicing [Krawczak et al., 1992]. Mutations in splice sites decrease recognition of the adjacent exon and consequently inhibit splicing of the adjacent intron [Talerico and Berget, 1990,Carothers et al., 1993]. Splice site mutations may result in exon skipping, activation of cryptic splice sites, creation of a pseudo-exon within an intron, or intron retention [Nakai and Sakamoto, 1994]: (1) Exon skipping, the most frequent outcome, is thought to result from failure of the normal and mutant splice sites to define an exon. (2) Most cryptic mutations activate splice sites of the same type and are typically located within a few hundred nucleotides of the natural site. This distance is probably limited by restrictions on the length of the resultant exon [Hawkins, 1988,Berget, 1995]. (3) Occasionally, mutations that are further away from the natural splice site create cryptic sites that are activated in the presence of a nearby cryptic splice site of opposite polarity, producing a novel non-coding exon within the intron. (4) Splice site mutations in very short or terminal introns can result in intron retention [Dominski and Kole, 1991]. In these instances, additional sequence elements may be required for normal splicing [Black, 1991,Black, 1992,Sterner and Berget, 1993].
Essential elements in donor and acceptor splice junctions have been defined by consensus sequences [Mount, 1982], by analysis of nucleotide frequencies at each position in a splice site [Senapathy et al., 1990] and by neural network prediction [Brunak et al., 1990]. Each of these methods have limitations. Although the GT and AG positions adjacent to donor and acceptor splice junctions are highly conserved, other positions are more variable [Mount, 1982,Stephens and Schneider, 1992]. The consensus sequence approximates the nucleotide frequencies at each position, and so it excludes the contributions of less frequent nucleotides present in a proportion of natural splice sites. Splice site sequences that deviate from the consensus do not necessarily produce significantly lower amounts of spliced mRNA [Rogan and Schneider, 1995]. Training a neural network requires sequences of both binding sites and sequences that are not bound [Stormo et al., 1982,Brunak et al., 1990]. Generally, non-bound sequences are taken to be those remaining after binding sites have been identified. However, these sequences do contain functional sites [Schneider, 1997b,Hengen et al., 1997], so neural networks may be inappropriately trained on overlapping data sets.
In contrast, information-theory based models of donor and acceptor splice sites require only functional sites and show which nucleotides are permissible at both highly-conserved and variable positions of these sites [Stephens and Schneider, 1992]. Information is the only measure of sequence conservation which is additive [Shannon, 1948]. The information content (Ri, in bits) of a member of a sequence family describes the degree to which that member contributes to the conservation of the entire family [Schneider, 1997a,Schneider, 1997b]. Ri is the dot product of a weight matrix derived from the nucleotide frequencies at each position of a splice site sequence database and the vector of a particular sequence. Individual information is related to thermodynamic entropy and therefore to the free energy of binding [Schneider, 1994,Schneider, 1997a]. Since splice sites are recognized prior to intron excision [Berget, 1995], the sequence of the splice site dictates the strength of the spliceosome-splice junction interaction and thus splice site use. It is our thesis that the strength of this interaction is related to the information content of the splice junction.
A group of sites with similar sequence and function can be described and
quantified by their corresponding distribution of individual information
contents. The mean of this distribution of Ri values is
![]() bits for the 10 nucleotide long splice donor sites and
bits for the 10 nucleotide long splice donor sites and
![]() bits for the 28 nucleotide long acceptor sequences
[Stephens and Schneider, 1992,Schneider, 1997a],
representing the average amount of
information required for splicing,
Rsequence[Schneider et al., 1986,Schneider, 1995,Schneider, 1994].
Strong splice sites have Ri values
bits for the 28 nucleotide long acceptor sequences
[Stephens and Schneider, 1992,Schneider, 1997a],
representing the average amount of
information required for splicing,
Rsequence[Schneider et al., 1986,Schneider, 1995,Schneider, 1994].
Strong splice sites have Ri values
![]() ;
weak sites have Ri values
;
weak sites have Ri values
![]() .
Non-functional sites have Rivalues less than or equal to zero [Schneider, 1994,Schneider, 1997a].
Since mutations at splice sites lessen or abolish splicing at those sites,
we investigated whether the Ri values of mutant splice
sites were related
to defects in mRNA processing and whether
mutant, cryptic and the corresponding natural splice sites could be ordered
based on their respective Ri values.
.
Non-functional sites have Rivalues less than or equal to zero [Schneider, 1994,Schneider, 1997a].
Since mutations at splice sites lessen or abolish splicing at those sites,
we investigated whether the Ri values of mutant splice
sites were related
to defects in mRNA processing and whether
mutant, cryptic and the corresponding natural splice sites could be ordered
based on their respective Ri values.
MATERIALS AND METHODS
Individual information analysis
Information content is defined as the number of choices needed to describe a
sequence pattern,
using a logarithmic scale in bits
[Schneider et al., 1986,Schneider, 1995].
A set of either donor or acceptor splice junction recognition sites are
aligned and the frequencies of bases at each position are determined.
The weight matrix used to model the splice junctions is computed from
The individual information of a sequence j is the dot product between the
sequence and the weight matrix:
The mean of the distribution of Ri values of natural sites is Rsequence [Schneider, 1997b,Schneider, 1997a]. The distribution of Ri values is approximately Gaussian, however the lower and upper bounds are zero bits and the Ri value of the consensus sequence.
The null Ri distribution was determined by creating a random
10,000 nucleotide sequence
with a Markov chain process that maintained the same mono- and dinucleotide
composition as the human splice junction database
[Stephens and Schneider, 1992].
The means of the splice donor and acceptor null distributions
were respectively
![]() and
and
![]() bits.
The probability of observing either
a donor or
acceptor site
with Ri> 0in this random sequence
was 0.02 (Z = 2.0).
bits.
The probability of observing either
a donor or
acceptor site
with Ri> 0in this random sequence
was 0.02 (Z = 2.0).
The effects of nucleotide substitutions
can be evaluated by comparing the individual
information
of the common and variant alleles.
The minimum fold change in binding affinity of two
sites is
![]() ,
where
,
where
![]() is the difference between
their respective individual information contents [Schneider, 1997a].
is the difference between
their respective individual information contents [Schneider, 1997a].
Computational tools
have been developed
to investigate and display individual information.
The
Riw(b,l)matrices were first computed from a set of 1799 splice donor and 1744 acceptor
sequences [Stephens and Schneider, 1992].
To scan for potential sites or
to determine the effects of a sequence change
on the normal and neighboring
sites, the individual information content of the donor or acceptor motif is
computed for every site-length window in the sequence.
To assess the effects of various substitutions on a specific
donor or acceptor site,
Ri was computed for the normal and variant sites
with the program
Scan
and displayed with
MakeWalker, DNAPlot and Lister
(Schneider.walker;
https://alum.mit.edu/www/toms/![]() toms/walker).
toms/walker).
The Scan program uses the Riw(b,l) matrix to evaluate the individual information (Ri) at each position in a sequence. For each evaluation, it also computes the number of standard deviations away from Rsequence (Z score), and the one-tailed probability (p) of observing a normal splice site with that value of Ri. Sequences with Ri values that are either significantly greater or less than Rsequencehave low probabilities of belonging to the natural population of sites.
A walker graphically shows the contributions of each position to a binding site. In the display (generated by Makewalker or Lister), favorable contacts between the spliceosome and a test sequence are indicated by letters that extend upwards, while positions that are predicted to make unfavorable contacts are shown by inverted letters. Makewalker is interactive and shows one walker at a time, while Lister displays multiple walkers aligned with sequences and annotated by coding regions (e.g. Figs 1-4).
Selection of mutations
Human splice site mutations were chosen from published reports for which corresponding genomic sequence data were available. Only a subset of reported mutations could be analyzed, as sufficient intron sequences were often unavailable (<26 nucleotides for acceptor sites, <7 nucleotides for donor sites). To investigate the relationship between Ri value and splice site use, studies that evaluated expression of the mutant mRNA were selected whenever possible. A sequence interval (>100 nucleotides) surrounding the splice junction was scanned to detect potential cryptic splice sites in the vicinity of the natural site. Larger sequence windows were used for cryptic sites known to occur further away from the natural site (e.g. Table 2, #24).
Two mutations could not be analyzed because there were discrepancies at corresponding splice site sequences from different reports. A mutation in the IVS 10 acceptor of the hexosaminidase B gene could not be analyzed because the natural acceptor site had negative information content in one of the sequences [Neote et al., 1988,Proia, 1988]. A similar inconsistency was found in two different versions of the IVS 5 acceptor sequence of the protein kinase C gene [Foster et al., 1985,Soria et al., 1993].
Statistical analyses
Natural and variant sites with Ri> 0were compared with Rsequence [Stephens and Schneider, 1992] by using the Z statistic and associated probability of observing a site with a particular Ri value [Schneider, 1997a].
Primary mutations
for either
donor or acceptor sites
were analyzed by determining
the average differences in
Ri values (
![]() )
of natural versus mutant
sequences.
Significance was evaluated using a paired t-test.
Mutations in which cryptic splicing
was either predicted or demonstrated experimentally
were excluded
to avoid biasing estimation of
)
of natural versus mutant
sequences.
Significance was evaluated using a paired t-test.
Mutations in which cryptic splicing
was either predicted or demonstrated experimentally
were excluded
to avoid biasing estimation of
![]() ,
since
cryptic splicing can alter natural splice site use
in the absence of a change in
the information content of that site.
,
since
cryptic splicing can alter natural splice site use
in the absence of a change in
the information content of that site.
The observed distributions of the locations of cryptic donor and acceptor sites were compared with a model that assumes that these sites are equally likely to occur upstream or downstream of the natural site. Significance was evaluated with the binomial distribution.
Relationship of information content to splice site use
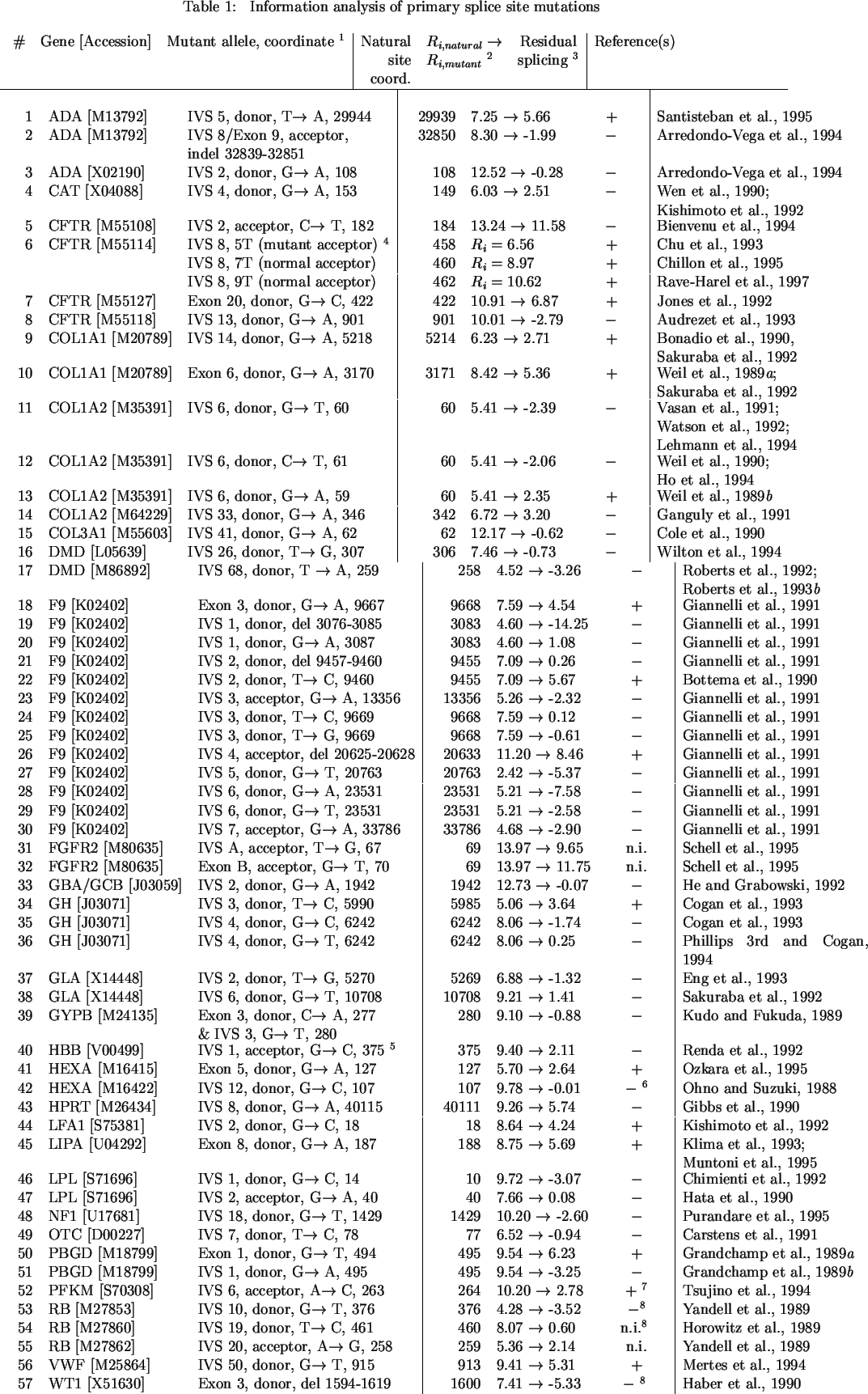
Different mutation reports measured splice site use directly by either cDNA sequencing, reverse transcription-PCR, primer-extension, S1 nuclease analyses, or allele-specific hybridization. Direct comparisons of natural and mutant splicing patterns were not always available. In some instances, the effect of the mutation was measured indirectly using either Northern hybridization (Table 1, #46, 47, 49; Table 3, #4), antigen immunoprecipation or protein levels (Table 1, #18, 19, 20, 21, 23, 24, 25, 26, 27, 28, 29, 30, 49; Table 2, #40, 41, 42, 43; Table 3, #2, 3, 4) or measurements of enzymatic activity (Table 1, #18, 19, 20, 21, 23, 24, 25, 26, 27, 28, 29, 30, 34, 35; Table 2, #40, 41, 42, 43 ; Table 3, #2, 3). Functional analyses of splicing were not reported for mutations #31, 32, 54 and 55 in Table 1, #14, 15, 23, 34-38, and 44, 45, 46 in Table 2, and #1, 7 and 8 (the natural site at 2621) in Table 3.
RESULTS
Several categories of mutations were distinguished by individual information analysis. A total of 111 nucleotide substitutions were evaluated. Fifty-seven mutations were nucleotide substitutions that solely altered use of the natural splice site and did not create cryptic splice sites (designated as primary splice site mutations, Table 1). Activated cryptic splice sites were predicted for 46 different mutations, 33 of which were corroborated experimentally (Table 2). Eight nucleotide substitutions were predicted not to alter splicing (Table 3).
I. Primary mutations in splice junction recognition sequences
Differences in information content of natural and mutant
splice sites.
Many of the primary splice junction mutants that showed complete exon
skipping
(residual splicing: -)
had Ri values ![]() bits
(Table 1, #2, 3, 11, 12, 15, 16, 17, 19, 35).
However, there are primary mutant donor and
acceptor sites that were not used
that have mostly small positive Ri values
(Table 1, #4, 5, 14, 20, 21, 24, 36, 38, 40, 43, 47).
This suggests that
recognition of splice donor and acceptor sites
requires more than zero bits.
bits
(Table 1, #2, 3, 11, 12, 15, 16, 17, 19, 35).
However, there are primary mutant donor and
acceptor sites that were not used
that have mostly small positive Ri values
(Table 1, #4, 5, 14, 20, 21, 24, 36, 38, 40, 43, 47).
This suggests that
recognition of splice donor and acceptor sites
requires more than zero bits.
Mutations that reduce or completely abolish splicing
have significantly lower Ri values than the corresponding
natural sites.
The average difference in Ri between primary mutant and natural
donor sites is
![]() bits (n = 45)
and for acceptor sites it is
bits (n = 45)
and for acceptor sites it is
![]() bits (n = 12),
and these differences are significant
(p<0.0001 for both
bits (n = 12),
and these differences are significant
(p<0.0001 for both
![]() values).
Ri values of
primary acceptor mutations range from
a minimum of -2.90 bits
to
a maximum of 11.75 bits,
whereas donor mutations have a lower range from
-14.25to
6.87bits.
values).
Ri values of
primary acceptor mutations range from
a minimum of -2.90 bits
to
a maximum of 11.75 bits,
whereas donor mutations have a lower range from
-14.25to
6.87bits.
We considered the possibility that the strength of a natural splice site,
i.e. Ri value,
might be related to its susceptibility to mutational
inactivation. 15 of 24 (62%) natural sites in Table 1
with Ri values
> Rsequencewere inactivated by mutation
or had mutant Ri values ![]() ,
compared to 22 of 29 (76%) natural sites with Ri values
< Rsequence.
Inactivation of splicing is primarily determined by the
specific nucleotide substitutions that occur at those sites,
however weak natural splice sites
may be more susceptible than strong sites to
succumb to
mutations that abolish splicing.
,
compared to 22 of 29 (76%) natural sites with Ri values
< Rsequence.
Inactivation of splicing is primarily determined by the
specific nucleotide substitutions that occur at those sites,
however weak natural splice sites
may be more susceptible than strong sites to
succumb to
mutations that abolish splicing.
Amount of information required for splicing. The minimum quantity of information required for splicing, Ri,min, was defined by comparing the Ri values of inactivating to leaky primary mutations (cryptic splicing mutations were excluded because activation of cryptic sites may affect natural site use). Ri,minis bounded by the maximum information content of a non-functional site and the minimum quantity of information required to produce normal transcripts.
The following minimally functional sites had small positive Ri values:
A mutation at the exon
5 donor site (
![]() bits)
in the HEXA gene results in a low level
(3%) of normal mRNA (Table 1, #41).
Similarly, a mutation at the exon 4 acceptor site
(
bits)
in the HEXA gene results in a low level
(3%) of normal mRNA (Table 1, #41).
Similarly, a mutation at the exon 4 acceptor site
(
![]() bits)
in the APOE gene results in 5% of
normal splicing
(Table 2, #2)
and
a mutation at the IVS 14 donor site (
bits)
in the APOE gene results in 5% of
normal splicing
(Table 2, #2)
and
a mutation at the IVS 14 donor site (
![]() bits)
in COL1A1 decreases
(by 50-60%)
but does not abolish normal splicing
(Table 1, #9).
Furthermore, a mutant 2.4 bit acceptor site in the IDS gene
(Table 2, #30) is associated with a moderately abnormal phenotype (the other
allele is null), consistent with production of some
normal mRNA.
Finally,
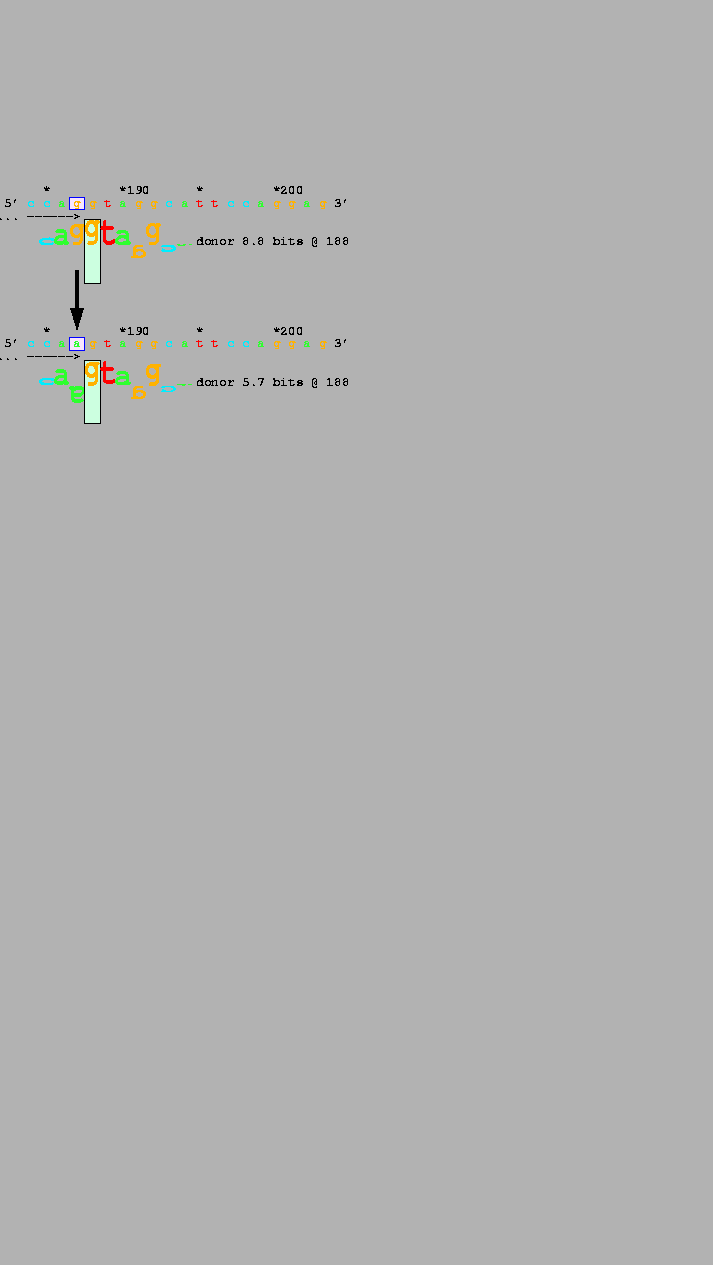
a mutation at the IVS 6 acceptor in COL1A2 reduces the Ri value of
the splice site from 5.4 to 2.4 bits and results in a mild form of
Ehlers-Danlos (type VII) syndrome due to 50% exon skipping (Table 1, #13;
Fig. 1).
Splicing at this site is completely impaired
in vitro at 39
bits)
in COL1A1 decreases
(by 50-60%)
but does not abolish normal splicing
(Table 1, #9).
Furthermore, a mutant 2.4 bit acceptor site in the IDS gene
(Table 2, #30) is associated with a moderately abnormal phenotype (the other
allele is null), consistent with production of some
normal mRNA.
Finally,
a mutation at the IVS 6 acceptor in COL1A2 reduces the Ri value of
the splice site from 5.4 to 2.4 bits and results in a mild form of
Ehlers-Danlos (type VII) syndrome due to 50% exon skipping (Table 1, #13;
Fig. 1).
Splicing at this site is completely impaired
in vitro at 39
![]() and
restored at 30
and
restored at 30
![]() .
The temperature sensitivity of this mutation indicates that this 2.4 bit
sequence is weakly bound by the spliceosome.
.
The temperature sensitivity of this mutation indicates that this 2.4 bit
sequence is weakly bound by the spliceosome.
By contrast, mutations
at the exon 1 donor splice site
in the CAT gene (Table 1, #4;
![]() bits),
in IVS 33 of COL1A2 (Table 1, #14;
bits),
in IVS 33 of COL1A2 (Table 1, #14;
![]() bits)
completely abolish mRNA splicing. The Ri value of this COL1A2 mutation
is inconsistent with the result found for mutation #13, since the mutation
with lower information content would be expected to be inactive.
This difference may
not be significant depending on the (unknown) precision of the
Riw(b,l)matrix, however it seems more likely that
residual splicing at the mutated site in mutation
#14 may not have been detected.
Residual
splicing was observed at several mutant splice sites with Ri values
greater than 2.4 bits
and less than 3.2 bits (Table 1, # 9, 41 and 52).
These splice junction mutations define a range of values
for Ri,min of either donor or acceptor sites.
Although the confidence interval around Ri,minis unknown, donor and acceptor splice sites with Ri>2.4 bits are
rarely found in a
set of random sequences with human dinucleotide composition (p=0.008).
To simplify comparisons between Ri,minand other Ri values,
we use
bits)
completely abolish mRNA splicing. The Ri value of this COL1A2 mutation
is inconsistent with the result found for mutation #13, since the mutation
with lower information content would be expected to be inactive.
This difference may
not be significant depending on the (unknown) precision of the
Riw(b,l)matrix, however it seems more likely that
residual splicing at the mutated site in mutation
#14 may not have been detected.
Residual
splicing was observed at several mutant splice sites with Ri values
greater than 2.4 bits
and less than 3.2 bits (Table 1, # 9, 41 and 52).
These splice junction mutations define a range of values
for Ri,min of either donor or acceptor sites.
Although the confidence interval around Ri,minis unknown, donor and acceptor splice sites with Ri>2.4 bits are
rarely found in a
set of random sequences with human dinucleotide composition (p=0.008).
To simplify comparisons between Ri,minand other Ri values,
we use
![]() bits.
bits.
Leaky splicing
To determine whether the information present in a mutant site
was related to splice site
use, the Ri values of mutated splice sites that
inactivated splicing were compared with
Ri values of leaky splice sites. Completely inactivated
sites generally had
Ri values less than Ri,min (e.g. Table 1, #46), whereas
mutations with Ri values greater than
Ri,min reduced but generally did not abolish splicing. For example,
a G
![]() C point mutation in the exon 2 donor site of the LFA1 gene
(Table 1, #44) decreased Ri from 8.6 to 4.2 bits and this
mutation is leaky, i.e. 3% of the normal spliced product
is detected from this allele
[Kishimoto et al., 1989].
Likewise, a
patient with mild cholesterol storage disease was homozygous for a donor site
mutation in the LIPA gene
(
C point mutation in the exon 2 donor site of the LFA1 gene
(Table 1, #44) decreased Ri from 8.6 to 4.2 bits and this
mutation is leaky, i.e. 3% of the normal spliced product
is detected from this allele
[Kishimoto et al., 1989].
Likewise, a
patient with mild cholesterol storage disease was homozygous for a donor site
mutation in the LIPA gene
(
![]() bits;
Table 1, #45; Fig. 2).
Mutations #1, 6, 7, 9, 10, 13, 18, 22, 26, 34, 41, 44, 45, 50, 52, and 56
(Table 1) and
#2, 3, 4, 7, 9, 16, 21,
23, 27, 28, 30, 32 and 41 (Table 2),
which have Ri values
bits;
Table 1, #45; Fig. 2).
Mutations #1, 6, 7, 9, 10, 13, 18, 22, 26, 34, 41, 44, 45, 50, 52, and 56
(Table 1) and
#2, 3, 4, 7, 9, 16, 21,
23, 27, 28, 30, 32 and 41 (Table 2),
which have Ri values
![]() ,
are leaky at the respective natural splice sites.
The average decrease in Ri values is smaller for
primary mutations that
result in reduced levels of
normally spliced mRNA;
,
are leaky at the respective natural splice sites.
The average decrease in Ri values is smaller for
primary mutations that
result in reduced levels of
normally spliced mRNA;
![]() is
is
![]() bits for donor sites (n = 12; versus -7.67 for all
donor sites)
and
bits for donor sites (n = 12; versus -7.67 for all
donor sites)
and
![]() for acceptor sites (n = 4; versus -5.97 for all
acceptor sites).
When cryptic splice site mutations that
result in residual splicing at the natural site
are considered in addition,
the change is negligible:
for acceptor sites (n = 4; versus -5.97 for all
acceptor sites).
When cryptic splice site mutations that
result in residual splicing at the natural site
are considered in addition,
the change is negligible:
![]() bits (n = 14) for
donor sites and
bits (n = 14) for
donor sites and
![]() bits (n = 15) for acceptor sites.
bits (n = 15) for acceptor sites.
Quantitative relationship
The quantitative relationship between splice site use
and information content is illustrated by the
polymorphic alleles in IVS 8 of the CFTR gene
(Table 1, #6; Fig. 3).
The frequency of exon 9 skipping is inversely
related to the length of the polypyrimidine tract of
the upstream acceptor site
[Chu et al., 1993,Chillon et al., 1995,Rave-Harel et al., 1997].
This is not surprising since
the length of a homopolymeric
polypyrimidine tract
has also been related to splice site strength [Dominski and Kole, 1991].
The 4.1 bit difference between
the Ri values of the shortest and longest alleles accounts for the
lower amount of spliced mRNA from the
shorter allele and is probably related to the phenotype
of congenital bilateral
absence of the vas deferens in male homozygotes.
A 4.1 bit reduction in information
would correspond to
at least a 17 fold
(
![]() )
decrease in splicing,
assuming minimal conversion of
information to energy dissipated
[Schneider, 1991b,Schneider, 1994].
This corresponds closely to
the relative amounts of mRNA produced by the
shortest (5T) and
longest (9T)
alleles [Chillon et al., 1995].
)
decrease in splicing,
assuming minimal conversion of
information to energy dissipated
[Schneider, 1991b,Schneider, 1994].
This corresponds closely to
the relative amounts of mRNA produced by the
shortest (5T) and
longest (9T)
alleles [Chillon et al., 1995].
Only two exceptional mutations were found in which
![]() ,
although
these sites
were reportedly not used (Table 1, #5 [11.6 bits], #43 [5.7 bits]).
The minimum predicted decreases
of
3 and 11 fold, respectively,
in binding affinity
would not be expected to completely abolish splicing at these sites.
Reduced amounts of splicing can
occur at mutant splice sites with
Ri> Ri,min, although a modest decrease in Ri at a splice site can
apparently sometimes inactivate splicing.
,
although
these sites
were reportedly not used (Table 1, #5 [11.6 bits], #43 [5.7 bits]).
The minimum predicted decreases
of
3 and 11 fold, respectively,
in binding affinity
would not be expected to completely abolish splicing at these sites.
Reduced amounts of splicing can
occur at mutant splice sites with
Ri> Ri,min, although a modest decrease in Ri at a splice site can
apparently sometimes inactivate splicing.
II. Detection of cryptic splice sites
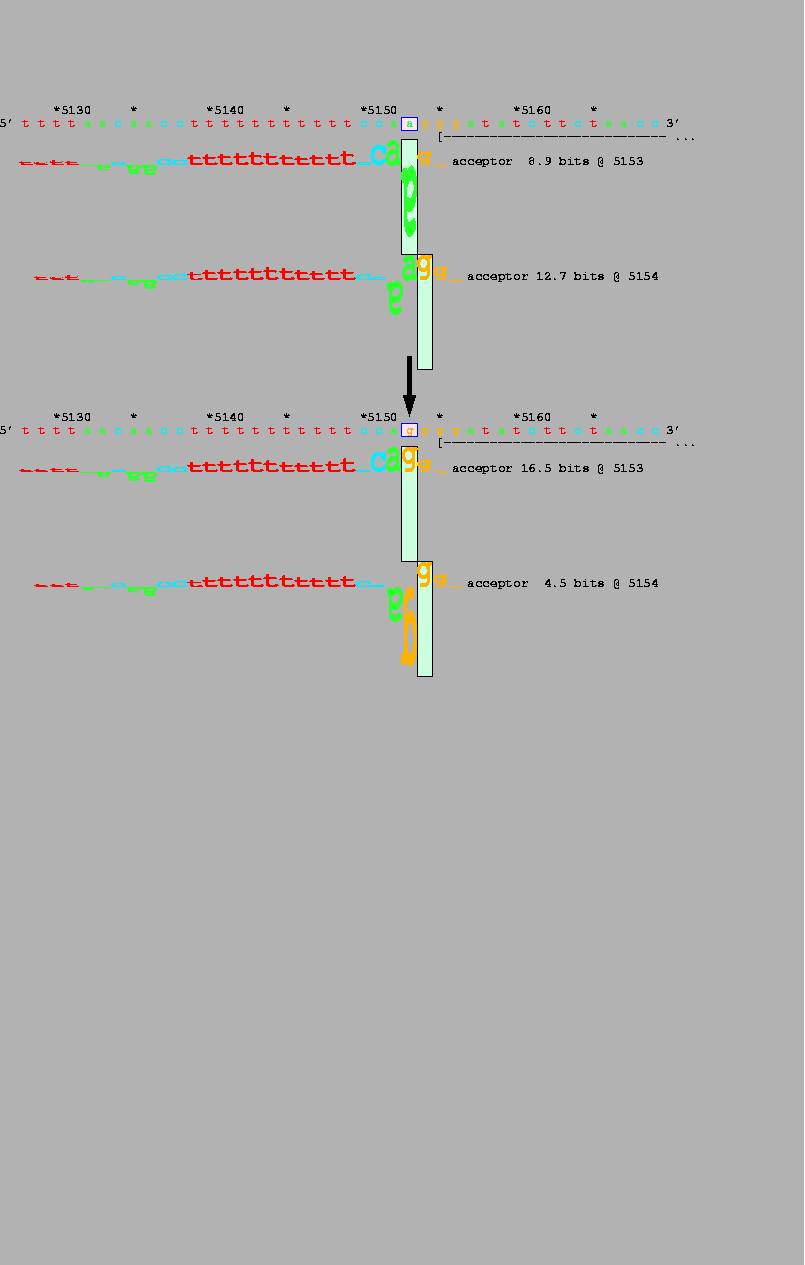
Categories of cryptic splice sites Ri analysis detected secondary cryptic splice sites that are activated by mutation in or adjacent to the natural primary splice site. This indicates that the Ri values of activated cryptic sites may be determined with an information model derived from natural splice sites [Stephens and Schneider, 1992]. Table 2 shows 33 experimentally-identified cryptic sites confirmed by information analysis of the respective genomic sequences (section A), and 13 mutations that were predicted by Ri analysis to exhibit cryptic splicing (section B). For example, a mutation at position 35066 of the adenosine deaminase gene (Table 2, #1) does not alter the Ri value of the natural splice site (at 35099), but creates a secondary cryptic site of similar strength at position 35067. There were 7 additional mutations in which a new cryptic site was either created or predicted without altering the Ri value of natural splice site (Table 2, #12, 14, 15, 26, 31, 40, 43). Activation of cryptic sites can also prevent splicing at natural sites by promoting exon skipping (e.g. in 79% of transcripts resulting from a mutation in the iduronate-2-sulfatase gene; Table 2, #26; [Jonsson et al., 1995]). Exon skipping mutations occurred predominantly at donor splice sites (7 of 8) and in each instance, a cryptic site was created upstream whose Ri value exceeded or was similar to that of the natural site.
Several types of cryptic splicing mutations were distinguished:
Susceptibility to activation
Of 31 experimentally-verified cryptic splicing mutations
(Table 2A, excluding #5 and 6),
there are 19 splice sites
whose Ri values exceeded the cryptic site
prior to its activation
(
![]() bits).
For the remaining 12 mutations
(10 of which involve the same site in HBB),
the inactive cryptic sites exceed
the natural site by only an
bits).
For the remaining 12 mutations
(10 of which involve the same site in HBB),
the inactive cryptic sites exceed
the natural site by only an
![]() of
of
![]() bits.
Furthermore,
the differences in Rivalues between natural and cryptic sites prior to mutational
activation are much
smaller for donor sites
(
bits.
Furthermore,
the differences in Rivalues between natural and cryptic sites prior to mutational
activation are much
smaller for donor sites
(
![]() ,
n = 17 for donors vs.
,
n = 17 for donors vs.
![]() ,
n = 15 for acceptors).
Likewise,
cryptic donors were activated by an increase of
,
n = 15 for acceptors).
Likewise,
cryptic donors were activated by an increase of
![]() bits (n=5),
whereas
cryptic acceptor sites were activated by
bits (n=5),
whereas
cryptic acceptor sites were activated by
![]() bits (n=10).
From these observations it would appear that
donor sites may be more susceptible to
the effects of neighboring cryptic sites.
bits (n=10).
From these observations it would appear that
donor sites may be more susceptible to
the effects of neighboring cryptic sites.
Distance effects
Cryptic sites activated by a mutation that weakens
the natural site must reside
within a few hundred nucleotides of the natural splice site, since
the novel exon is restricted in length
[Hawkins, 1988,Berget, 1995].
For example, a strong cryptic acceptor in intron 2 of the
![]() -hemoglobin gene is
activated by mutations at the exon 3 acceptor 271 nucleotides downstream
(Table 2, #24).
Mutation at a natural site can, however, activate sites that are
further away when a cryptic exon is created.
For example,
mutation at the exon 3 acceptor of the CFTR gene activates
a cryptic, non-coding exon
in intron 3 (2,354 nucleotides downstream of exon 3 and 19,329
nucleotides upstream of exon 4;
Table 2, #3).
-hemoglobin gene is
activated by mutations at the exon 3 acceptor 271 nucleotides downstream
(Table 2, #24).
Mutation at a natural site can, however, activate sites that are
further away when a cryptic exon is created.
For example,
mutation at the exon 3 acceptor of the CFTR gene activates
a cryptic, non-coding exon
in intron 3 (2,354 nucleotides downstream of exon 3 and 19,329
nucleotides upstream of exon 4;
Table 2, #3).
Exceptions
Although pre-existing or novel cryptic sites with Ri values
less than that of the strongest local splice site were usually
not recognized, there were exceptions.
Infrequently, a weaker cryptic site can interfere with a natural
site, even when the natural site is strengthened by the mutation
(e.g. Table 2, #16).
For example,
activated cryptic sites with Ri values lower than those of the
natural splice site after mutation
may sometimes be used
(Table 2, #1, 3, 4, 6, 9, 16, 23, 32).
In at least one instance
(Table 2, #1),
a cryptic acceptor site upstream of the natural site is
predominantly used
despite the fact that both sites
have similar Rivalues, which suggests that the cryptic site is recognized first.
Conversely, the Ri value of the exon 1 donor
in the ![]() -globin gene
is less than that of an upstream cryptic site
(Table 2, #12-15, 17-22),
however this cryptic site is not activated unless
it is strengthened
or
the donor is weakened.
These exceptions suggest that
besides direct
competition between the cryptic and
natural splice sites,
other factors can influence splice site selection.
-globin gene
is less than that of an upstream cryptic site
(Table 2, #12-15, 17-22),
however this cryptic site is not activated unless
it is strengthened
or
the donor is weakened.
These exceptions suggest that
besides direct
competition between the cryptic and
natural splice sites,
other factors can influence splice site selection.
Another class of exceptional splice sites were those that generated alternatively processed transcripts. Active ``cryptic'' sites that resided in introns of the CSPB gene had Ri values in the normal range (Table 2, #5, 6) [Trapani et al., 1988,Klein et al., 1989]. They may represent alternative splice sites regulated by other sequence elements that can be present in the adjacent exons [Lavigueur et al., 1993,Sun et al., 1993b,Dirksen et al., 1994,Huh and Hynes, 1994,Humphrey et al., 1995] or polypyrimidine tracts [Sun et al., 1993b,Wang et al., 1995].
III. Non-deleterious splice site substitutions
Nucleotide substitutions that do not significantly alter the Ri value of a natural site are expected to produce functional rather than mutant sites [Rogan and Schneider, 1995]. Given that such substitutions are not likely to be deleterious, they may be polymorphic in the germline, as has been shown for a sequence change in an hMSH2 splice acceptor site [Leach et al., 1993]. We identified other nucleotide substitutions that did not significantly alter the Ri value (Table 3):
Splicing patterns for several nucleotide substitutions #1, 2, 3, and 7 (Table 3) were not reported, however, based on information analysis, these changes would not be predicted to alter mRNA splicing. The substitutions either maintain or increase the information content of the natural splice site. The Ri values of the proposed cryptic sites for substitutions #1, 2, and 8 were either negative or unchanged, suggesting that they are not activated by these substitutions. A proposed cryptic site in exon 3 of the p53 gene (substitution #7) is significantly weaker than the natural acceptor site (by 6.14 bits) and has an Ri value only slightly larger than Ri,min. It would seem unlikely that this cryptic site is preferentially used.
DISCUSSION
The number of bits in a splice site is related to the amount of splicing at that site. Previously, we demonstrated that a polymorphic splice junction variant caused little change in information [Rogan and Schneider, 1995]. The present study extends this finding and shows that mutant splice sites often contain significantly less information than their corresponding natural sites. Further, cryptic splice sites are activated by increases in information or by decreases at the natural splice site, and the information at activated cryptic sites is often comparable to or exceeds the natural site.
Predicting the effects of mutations A required step of information analysis is to compute the total information over all positions in a site. This value must then be compared with that of other sites prior to concluding that a substitution that changes a positive to a negative weighting is deleterious (compare Tables 1 and 2 to Table 3). Functional splice sites can have nucleotides with negative weightings (e.g. Fig. 1, position 63) that are offset by strong contributions at other positions (e.g. Fig. 1, position 64), as we have shown for other binding sites (Figure 2 in Schneider.walker, Hengen.fisinfo). Statistical analyses of the distributions of point mutations in splice sites are useful [Krawczak et al., 1992] but can sometimes obscure these compensating effects. Within a binding site, the context of a mutation can be as important as the mutation itself.
The difference between the observed value of Ri,min (![]() bits) and its
expected value (zero bits) may have a biological basis.
However,
this difference could also be explained by errors
in the database used to create the
splice weight matrices [Schneider, 1997b],
statistical limitations of the data and matrices,
motifs that are different from the
majority of sites [Hall and Padgett, 1994],
or
intrinsic limits to the precision of splice site recognition
[Schneider, 1991a].
Although the standard deviation of
Rsequence can be determined [Stephens and Schneider, 1992],
the confidence intervals on individual Rivalues are unknown.
These intervals are expected to be larger
at the
lower
and
upper
bounds of the Ri distribution,
where fewer functional splice
sites are observed.
The existence of a natural site with
Ri< Ri,min
(2.2 bits; Table 2, #26)
and an exon-skipping mutation with
Ri> Ri,min
(3.2 bits; Table 1, #14)
suggests that
Ri,min is not known precisely.
The error
(
|Ri- Ri,min|)
may be
as little as 0.2 bits
(
Ri= 2.2 bits; Table 2, #26),
but it might be
as much as
2.4 bits
(Ri= 0 bits; Schneider.Ri).
bits) and its
expected value (zero bits) may have a biological basis.
However,
this difference could also be explained by errors
in the database used to create the
splice weight matrices [Schneider, 1997b],
statistical limitations of the data and matrices,
motifs that are different from the
majority of sites [Hall and Padgett, 1994],
or
intrinsic limits to the precision of splice site recognition
[Schneider, 1991a].
Although the standard deviation of
Rsequence can be determined [Stephens and Schneider, 1992],
the confidence intervals on individual Rivalues are unknown.
These intervals are expected to be larger
at the
lower
and
upper
bounds of the Ri distribution,
where fewer functional splice
sites are observed.
The existence of a natural site with
Ri< Ri,min
(2.2 bits; Table 2, #26)
and an exon-skipping mutation with
Ri> Ri,min
(3.2 bits; Table 1, #14)
suggests that
Ri,min is not known precisely.
The error
(
|Ri- Ri,min|)
may be
as little as 0.2 bits
(
Ri= 2.2 bits; Table 2, #26),
but it might be
as much as
2.4 bits
(Ri= 0 bits; Schneider.Ri).
Susceptibility to mutation Donor sites may be more susceptible to inactivation than acceptor sites. The Ri values of mutant donor sites are more likely than mutant acceptors to be less than Ri,min. Natural donors possess less information than acceptors [Stephens and Schneider, 1992] and the average decrease in information due to mutation at donor sites exceeds the reduction in Ri at acceptors. Information is also less densely distributed across acceptor splice sites (0.3 bits per nucleotide) than in donor sites (0.8 bits per nucleotide), so changes at acceptors often have a smaller effect on Ri. Significantly more primary mutations in donor sites (n=45) than acceptor sites (n=12) were found, as has been noted [Krawczak et al., 1992,Nakai and Sakamoto, 1994].
Cryptic splicing The Ri values of most novel cryptic donor sites exceeded or were similar to those of the corresponding natural sites. Although similar results were also inferred from Shapiro-Senapathy consensus values [Krawczak et al., 1992], information analysis detects fewer incorrect cryptic splice sites [O'Neill et al., 1998], more accurately discriminates true sites from non-sites, and visually depicts both changes (Fig. 4).
An exon is initially defined by recognizing the acceptor [Berget, 1995]. Cryptic acceptor sites occur either upstream (n = 9) or downstream (n = 7) of the natural site (p = 0.4), suggesting that they are not located by scanning [Stephens and Schneider, 1992]. The exon definition model predicts that the spliceosome then scans downstream until a strong donor site is located [Robberson et al., 1990,Niwa et al., 1992], so a novel cryptic donor site created downstream of an intact natural site should not be recognized unless the natural site is mutated. In all cases, a decrease in the information content of the natural donor site activated pre-existing cryptic sites downstream (Table 2A). Furthermore, cryptic donor sites were activated more frequently upstream of the natural site (15 of 20; p=0.02). The idea that the splicing machinery selects for the strongest local acceptor splice site and scans for donors is supported by Ri analysis.
Nucleotide substitutions within 17 natural acceptor sites have been shown to create or strengthen adjacent cryptic sites that are thereby activated (see results: II. Detection of cryptic splice sites). Only acceptors were found, perhaps because the variable polypyrimidine tract potentiates spliceosome recognition at many positions, whereas donor sites have high information density and a non-repeating sequence pattern [Stephens and Schneider, 1992]. For this reason, weaker cryptic sites are often found near natural acceptor sites (e.g Fig. 4). Mutations involving the natural acceptor sometimes strengthen and activate these cryptic sites. The resulting aberrant exons may in some cases have been misidentified as natural splice products (e.g. Table 2B), since their length and sequence would differ by only a few nucleotides from the normal mRNA.
Conclusion We have shown that individual information theory can be used to rank normal and mutant splice junctions. As a consequence, silent polymorphisms can be distinguished from true mutations, changes in individual information are related to splice site use, and activated cryptic splice sites can be detected. These distinctions are possible because the information measure is related to the thermodynamic entropy, and therefore can be connected to the binding energy [Szilard, 1964,Schneider, 1991a,Schneider, 1991b,Schneider, 1994]. The information in the splice site should be related to the specific binding interaction between the spliceosome and the site [Berg and von Hippel, 1987,Berg and von Hippel, 1988a,Berg, 1988,Berg and von Hippel, 1988b]. However, the relationship is an inequality--the second law of thermodynamics [Schneider, 1991b,Schneider, 1994]--and can only be explored empirically at this stage. The correlation between information measures and measured thermodynamic parameters is expected to more precisely relate genotypes to phenotypes in genetic disorders.
ACKNOWLEDGEMENTS
We thank Greg Alvord for statistical consulting, and Kenn Kraemer and Howard Young for reading the manuscript. Grant support is acknowledged from the Public Health Service (CA74683) and the American Cancer Society (DHP-132) to P.K.R. We thank the Frederick Biomedical Supercomputing Center for access to computer resources and support services.
21
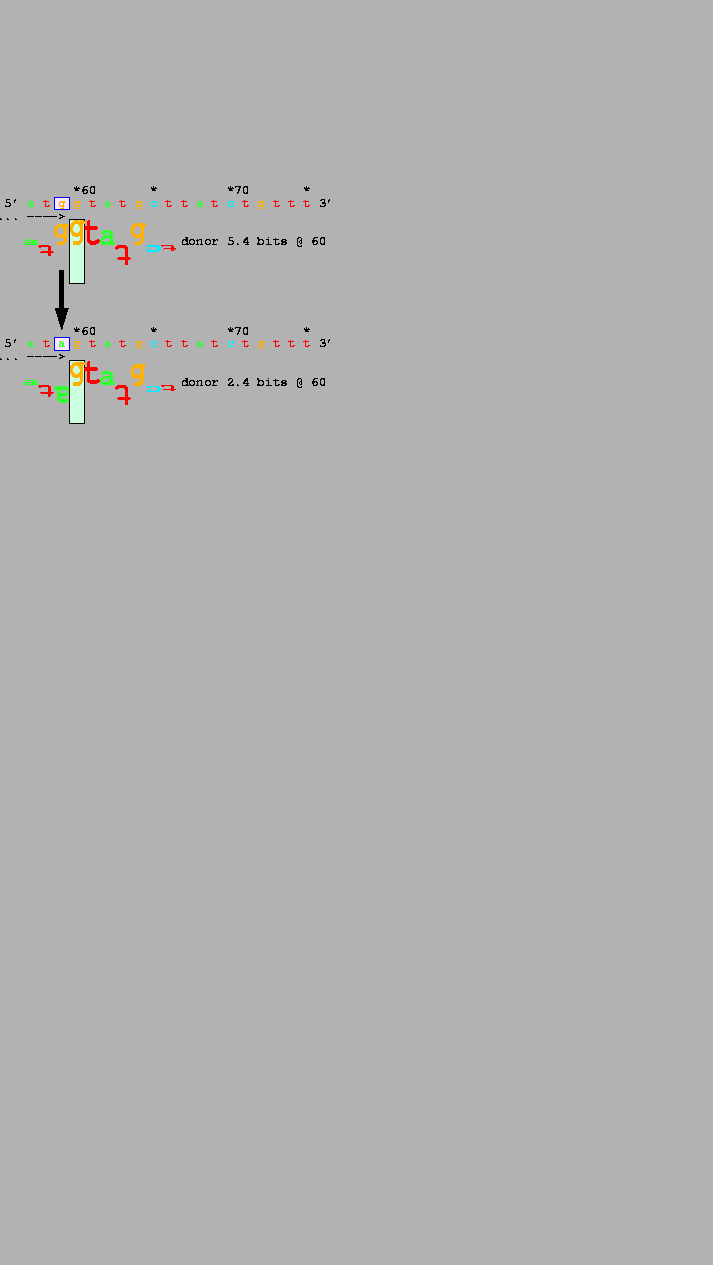
A G |
A G |
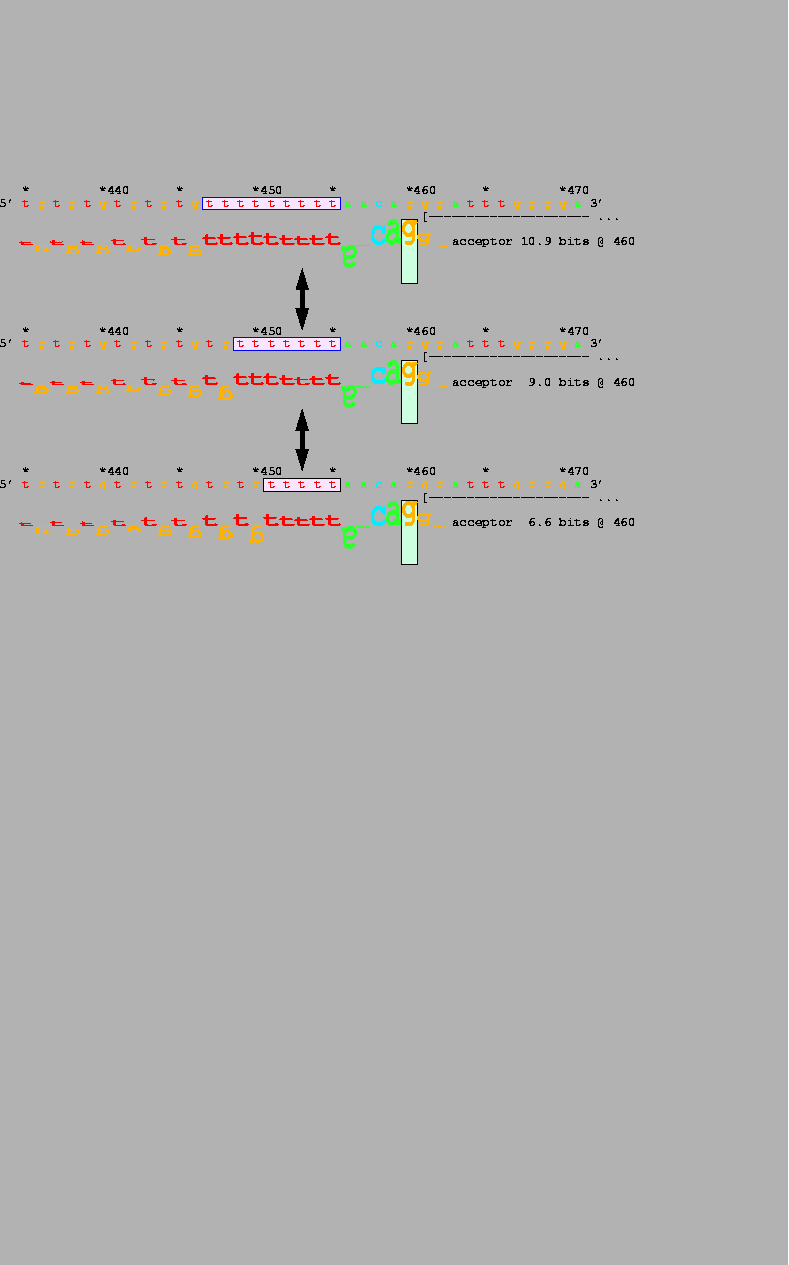
Splicing varies among 3 common alleles that differ in length in the polymorphic polythymidine tract of the IVS 8 acceptor of the gene encoding the cystic fibrosis transmembrane regulator [CFTR; M55114] (Table 1, #6). The shortest allele (bottom walker) shows 90% outsplicing of exon 9 and is associated with congenital absence of the vas deferens. Individuals with the two longer alleles have a normal phenotype, although the 7T allele produces less mRNA than the 9T allele. Exon 9 begins at the base indicated by the left bracket and dashes. |
An A |